| 登録情報 | データベース: PDB / ID: 4gwt
|
|---|
| タイトル | Structure of racemic Pin1 WW domain cocrystallized with DL-malic acid |
|---|
 要素 要素 | Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 |
|---|
 キーワード キーワード | PROTEIN BINDING / racemic crystallization / WW domain / proline phosphoSer/Thr binding |
|---|
| 機能・相同性 |  機能・相同性情報 機能・相同性情報
cis-trans isomerase activity / phosphothreonine residue binding / negative regulation of cell motility / ubiquitin ligase activator activity / regulation of protein localization to nucleus / GTPase activating protein binding / mitogen-activated protein kinase kinase binding / protein targeting to mitochondrion / protein peptidyl-prolyl isomerization / regulation of mitotic nuclear division ...cis-trans isomerase activity / phosphothreonine residue binding / negative regulation of cell motility / ubiquitin ligase activator activity / regulation of protein localization to nucleus / GTPase activating protein binding / mitogen-activated protein kinase kinase binding / protein targeting to mitochondrion / protein peptidyl-prolyl isomerization / regulation of mitotic nuclear division / negative regulation of SMAD protein signal transduction / PI5P Regulates TP53 Acetylation / negative regulation of amyloid-beta formation / cytoskeletal motor activity / RHO GTPases Activate NADPH Oxidases / phosphoserine residue binding / postsynaptic cytosol / negative regulation of protein binding / Rho protein signal transduction / regulation of cytokinesis / Negative regulators of DDX58/IFIH1 signaling / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / phosphoprotein binding / negative regulation of transforming growth factor beta receptor signaling pathway / synapse organization / beta-catenin binding / negative regulation of protein catabolic process / regulation of protein stability / negative regulation of ERK1 and ERK2 cascade / ISG15 antiviral mechanism / tau protein binding / positive regulation of protein phosphorylation / neuron differentiation / positive regulation of canonical Wnt signaling pathway / regulation of gene expression / midbody / cellular response to hypoxia / Regulation of TP53 Activity through Phosphorylation / response to hypoxia / protein stabilization / nuclear speck / ciliary basal body / glutamatergic synapse / positive regulation of transcription by RNA polymerase II / nucleoplasm / nucleus / cytosol / cytoplasm類似検索 - 分子機能 : / Peptidyl-prolyl cis-trans isomerase, PpiC-type, conserved site / PpiC-type peptidyl-prolyl cis-trans isomerase signature. / PPIC-type PPIASE domain / PpiC-type peptidyl-prolyl cis-trans isomerase family profile. / Peptidyl-prolyl cis-trans isomerase, PpiC-type / WW domain / WW/rsp5/WWP domain signature. / WW domain superfamily / WW/rsp5/WWP domain profile. ...: / Peptidyl-prolyl cis-trans isomerase, PpiC-type, conserved site / PpiC-type peptidyl-prolyl cis-trans isomerase signature. / PPIC-type PPIASE domain / PpiC-type peptidyl-prolyl cis-trans isomerase family profile. / Peptidyl-prolyl cis-trans isomerase, PpiC-type / WW domain / WW/rsp5/WWP domain signature. / WW domain superfamily / WW/rsp5/WWP domain profile. / Domain with 2 conserved Trp (W) residues / WW domain / Peptidyl-prolyl cis-trans isomerase domain superfamily類似検索 - ドメイン・相同性 (2S)-2-hydroxybutanedioic acid / Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1類似検索 - 構成要素 |
|---|
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) |
|---|
| 手法 |  X線回折 / 分子置換 / 解像度: 2.25 Å X線回折 / 分子置換 / 解像度: 2.25 Å |
|---|
 データ登録者 データ登録者 | Mortenson, D.E. / Yun, H.G. / Gellman, S.H. / Forest, K.T. |
|---|
 引用 引用 | ジャーナル: Acta Crystallogr.,Sect.D / 年: 2013
タイトル: Evidence for small-molecule-mediated loop stabilization in the structure of the isolated Pin1 WW domain.
著者: Mortenson, D.E. / Kreitler, D.F. / Yun, H.G. / Gellman, S.H. / Forest, K.T. |
|---|
| 履歴 | | 登録 | 2012年9月3日 | 登録サイト: RCSB / 処理サイト: RCSB |
|---|
| 改定 1.0 | 2013年10月16日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2014年1月8日 | Group: Database references |
|---|
| 改定 1.2 | 2017年11月15日 | Group: Refinement description / カテゴリ: software
Item: _software.classification / _software.contact_author ..._software.classification / _software.contact_author / _software.contact_author_email / _software.date / _software.language / _software.location / _software.name / _software.type / _software.version |
|---|
| 改定 1.3 | 2023年9月13日 | Group: Data collection / Database references / Refinement description
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / struct_ref_seq_dif
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_ref_seq_dif.details |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード






 PDBj
PDBj







 集合体
集合体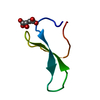
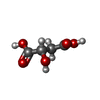
 分子量: 134.087 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C4H6O5
分子量: 134.087 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C4H6O5 分子量: 18.015 Da / 分子数: 11 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 11 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析