| Entry | Database: PDB / ID: 4gwv
|
|---|
| Title | Structure of racemic Pin1 WW domain cocrystallized with tri-ammonium citrate |
|---|
 Components Components | Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 |
|---|
 Keywords Keywords | PROTEIN BINDING / racemic crystallization / proline phosphoSer/Thr binding |
|---|
| Function / homology |  Function and homology information Function and homology information
cis-trans isomerase activity / phosphothreonine residue binding / negative regulation of cell motility / regulation of protein localization to nucleus / negative regulation of brown fat cell differentiation / mitogen-activated protein kinase kinase binding / ubiquitin ligase activator activity / GTPase activating protein binding / : / protein peptidyl-prolyl isomerization ...cis-trans isomerase activity / phosphothreonine residue binding / negative regulation of cell motility / regulation of protein localization to nucleus / negative regulation of brown fat cell differentiation / mitogen-activated protein kinase kinase binding / ubiquitin ligase activator activity / GTPase activating protein binding / : / protein peptidyl-prolyl isomerization / regulation of mitotic nuclear division / negative regulation of SMAD protein signal transduction / PI5P Regulates TP53 Acetylation / negative regulation of amyloid-beta formation / cytoskeletal motor activity / phosphoserine residue binding / RHO GTPases Activate NADPH Oxidases / postsynaptic cytosol / Rho protein signal transduction / regulation of cytokinesis / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / Negative regulators of DDX58/IFIH1 signaling / negative regulation of transforming growth factor beta receptor signaling pathway / regulation of protein stability / phosphoprotein binding / negative regulation of ERK1 and ERK2 cascade / negative regulation of protein catabolic process / positive regulation of protein phosphorylation / synapse organization / beta-catenin binding / protein destabilization / tau protein binding / ISG15 antiviral mechanism / neuron differentiation / positive regulation of canonical Wnt signaling pathway / regulation of gene expression / midbody / cellular response to hypoxia / Regulation of TP53 Activity through Phosphorylation / response to hypoxia / nuclear speck / ciliary basal body / protein stabilization / glutamatergic synapse / positive regulation of transcription by RNA polymerase II / nucleoplasm / nucleus / cytosol / cytoplasmSimilarity search - Function : / Peptidyl-prolyl cis-trans isomerase, PpiC-type, conserved site / PpiC-type peptidyl-prolyl cis-trans isomerase signature. / PPIC-type PPIASE domain / PpiC-type peptidyl-prolyl cis-trans isomerase family profile. / Peptidyl-prolyl cis-trans isomerase, PpiC-type / WW domain / WW/rsp5/WWP domain signature. / WW domain superfamily / WW/rsp5/WWP domain profile. ...: / Peptidyl-prolyl cis-trans isomerase, PpiC-type, conserved site / PpiC-type peptidyl-prolyl cis-trans isomerase signature. / PPIC-type PPIASE domain / PpiC-type peptidyl-prolyl cis-trans isomerase family profile. / Peptidyl-prolyl cis-trans isomerase, PpiC-type / WW domain / WW/rsp5/WWP domain signature. / WW domain superfamily / WW/rsp5/WWP domain profile. / Domain with 2 conserved Trp (W) residues / WW domain / Peptidyl-prolyl cis-trans isomerase domain superfamilySimilarity search - Domain/homology |
|---|
| Biological species |  Homo sapiens (human) Homo sapiens (human) |
|---|
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 3.05 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 3.05 Å |
|---|
 Authors Authors | Mortenson, D.E. / Yun, H.G. / Gellman, S.H. / Forest, K.T. |
|---|
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2013
Title: Evidence for small-molecule-mediated loop stabilization in the structure of the isolated Pin1 WW domain.
Authors: Mortenson, D.E. / Kreitler, D.F. / Yun, H.G. / Gellman, S.H. / Forest, K.T. |
|---|
| History | | Deposition | Sep 3, 2012 | Deposition site: RCSB / Processing site: RCSB |
|---|
| Revision 1.0 | Oct 16, 2013 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | Jan 8, 2014 | Group: Database references |
|---|
| Revision 1.2 | Nov 15, 2017 | Group: Refinement description / Category: software
Item: _software.classification / _software.contact_author ..._software.classification / _software.contact_author / _software.contact_author_email / _software.date / _software.language / _software.location / _software.name / _software.type / _software.version |
|---|
| Revision 1.3 | Feb 28, 2024 | Group: Data collection / Database references
Category: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / struct_ref_seq_dif
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _struct_ref_seq_dif.details |
|---|
|
|---|
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj












 Assembly
Assembly
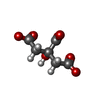
 Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7
Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 18.015 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing