- PDB-4g08: Crystal structure of the periplasmic domain of InvG -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4g08
Title
Crystal structure of the periplasmic domain of InvG
Components
Protein InvG
Keywords
CELL INVASION / Ring-building motif / protein secretion / PrgH
Function / homology
Function and homology information
type III protein secretion system complex / type II protein secretion system complex / protein secretion by the type III secretion system / protein secretion / cell outer membrane / identical protein binding Similarity search - Function
Ribosomal Protein S8; Chain: A, domain 1 - #120 / Phage tail protein beta-alpha-beta fold - #30 / Phage tail protein beta-alpha-beta fold / : / SPI-1 type 3 secretion system secretin, N0 domain / Type III secretion system outer membrane pore YscC/HrcC / : / Bacterial type II secretion system protein D signature. / Type II secretion system protein GspD, conserved site / NolW-like ...Ribosomal Protein S8; Chain: A, domain 1 - #120 / Phage tail protein beta-alpha-beta fold - #30 / Phage tail protein beta-alpha-beta fold / : / SPI-1 type 3 secretion system secretin, N0 domain / Type III secretion system outer membrane pore YscC/HrcC / : / Bacterial type II secretion system protein D signature. / Type II secretion system protein GspD, conserved site / NolW-like / NolW-like superfamily / Bacterial type II/III secretion system short domain / Type II/III secretion system / Bacterial type II and III secretion system protein / 3-Layer(bab) Sandwich / Ribosomal Protein S8; Chain: A, domain 1 / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology
Journal: PLoS Pathog / Year: 2013 Title: A refined model of the prototypical Salmonella SPI-1 T3SS basal body reveals the molecular basis for its assembly. Authors: Julien R C Bergeron / Liam J Worrall / Nikolaos G Sgourakis / Frank DiMaio / Richard A Pfuetzner / Heather B Felise / Marija Vuckovic / Angel C Yu / Samuel I Miller / David Baker / Natalie C J Strynadka / Abstract: The T3SS injectisome is a syringe-shaped macromolecular assembly found in pathogenic Gram-negative bacteria that allows for the direct delivery of virulence effectors into host cells. It is composed ...The T3SS injectisome is a syringe-shaped macromolecular assembly found in pathogenic Gram-negative bacteria that allows for the direct delivery of virulence effectors into host cells. It is composed of a "basal body", a lock-nut structure spanning both bacterial membranes, and a "needle" that protrudes away from the bacterial surface. A hollow channel spans throughout the apparatus, permitting the translocation of effector proteins from the bacterial cytosol to the host plasma membrane. The basal body is composed largely of three membrane-embedded proteins that form oligomerized concentric rings. Here, we report the crystal structures of three domains of the prototypical Salmonella SPI-1 basal body, and use a new approach incorporating symmetric flexible backbone docking and EM data to produce a model for their oligomeric assembly. The obtained models, validated by biochemical and in vivo assays, reveal the molecular details of the interactions driving basal body assembly, and notably demonstrate a conserved oligomerization mechanism.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Salmonella enterica subsp. enterica serovar Typhimurium (bacteria)
Salmonella enterica subsp. enterica serovar Typhimurium (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj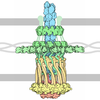
 Assembly
Assembly
 Mass: 18.015 Da / Num. of mol.: 112 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 112 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing