 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4fsr | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the CHK1 | ||||||
 Components Components | Serine/threonine-protein kinase Chk1 | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE inhibitor / TRANSFERASE / TRANSFERASE-TRANSFERASE inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of mitotic nuclear division / apoptotic process involved in development / negative regulation of G0 to G1 transition / histone H3T11 kinase activity / regulation of mitotic centrosome separation / mitotic G2/M transition checkpoint / inner cell mass cell proliferation / nucleus organization / regulation of double-strand break repair via homologous recombination / peptidyl-threonine phosphorylation ...negative regulation of mitotic nuclear division / apoptotic process involved in development / negative regulation of G0 to G1 transition / histone H3T11 kinase activity / regulation of mitotic centrosome separation / mitotic G2/M transition checkpoint / inner cell mass cell proliferation / nucleus organization / regulation of double-strand break repair via homologous recombination / peptidyl-threonine phosphorylation / mitotic G2 DNA damage checkpoint signaling / negative regulation of gene expression, epigenetic / Transcriptional Regulation by E2F6 / replicative senescence / Presynaptic phase of homologous DNA pairing and strand exchange / Activation of ATR in response to replication stress / Chk1/Chk2(Cds1) mediated inactivation of Cyclin B:Cdk1 complex / signal transduction in response to DNA damage / DNA damage checkpoint signaling / positive regulation of cell cycle / regulation of signal transduction by p53 class mediator / condensed nuclear chromosome / replication fork / TP53 Regulates Transcription of DNA Repair Genes / cellular response to mechanical stimulus / Signaling by SCF-KIT / G2/M DNA damage checkpoint / Ubiquitin-Mediated Degradation of Phosphorylated Cdc25A / G2/M transition of mitotic cell cycle / regulation of cell population proliferation / Processing of DNA double-strand break ends / Regulation of TP53 Activity through Phosphorylation / protein phosphorylation / protein kinase activity / non-specific serine/threonine protein kinase / DNA replication / chromatin remodeling / protein domain specific binding / protein serine kinase activity / DNA repair / protein serine/threonine kinase activity / apoptotic process / centrosome / DNA damage response / chromatin / protein-containing complex / : / nucleoplasm / ATP binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Kang, Y.N. / Stuckey, J.A. / Chang, P. / Russell, A.J. | ||||||
 Citation Citation | Journal: To be Published Title: Crystal Structure of the CHK1 Authors: Kang, Y.N. / Stuckey, J.A. / Chang, P. / Russell, A.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4fsr.cif.gz | 124 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4fsr.ent.gz | 95.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4fsr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fs/4fsrftp://data.pdbj.org/pub/pdb/validation_reports/fs/4fsr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4fsmC  4fsnC  4fsqC  4fstC  4fsuC  4fswC  4fsyC  4fszC  4ft0C  4ft3C  4ft5C  4ft7C  4ft9C  4ftaC  4ftcC  4ftiC  4ftjC  4ftkC  4ftlC  4ftmC  4ftnC  4ftoC  4ftqC  4ftrC  4fttC  4ftuC  4gh2C  4ft1 C: citing same article ( |
|---|---|
| Similar structure data |


















-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | biological unit is the same as asym. |
-Components
| #1: Protein | Mass: 32154.049 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Strain: human / Gene: CHEK1, CHK1 / Production host: HOMO SAPIENS (human)References: UniProt: O14757, non-specific serine/threonine protein kinase |
|---|---|
| #2: Chemical | ChemComp-HKC / 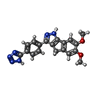  Mass: 360.369 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H16N6O2 Mass: 360.369 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H16N6O2 |
| #3: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 134 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 134 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.67 Å3/Da / Density % sol: 53.92 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: PEG 8000, Isopropanol, HEPES, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 298.0K |
-Data collection
| Diffraction | Mean temperature: 110 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.54 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Details: mirrors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: Osmic Si / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.5→20 Å / Num. all: 12003 / Num. obs: 11667 / % possible obs: 97.2 % / Observed criterion σ(I): -3 / Rmerge(I) obs: 0.085 / Χ2: 1.104 / Net I/σ(I): 9.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.5→19.7 Å / Cor.coef. Fo:Fc: 0.9391 / Cor.coef. Fo:Fc free: 0.9383 / Occupancy max: 1 / Occupancy min: 0 / SU R Cruickshank DPI: 0.429 / Cross valid method: THROUGHOUT / σ(F): 0
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 129.88 Å2 / Biso mean: 36.9651 Å2 / Biso min: 9.79 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.262 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→19.7 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.49→2.73 Å / Total num. of bins used: 6
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
