negative regulation of mitotic nuclear division / apoptotic process involved in development / negative regulation of G0 to G1 transition / histone H3T11 kinase activity / regulation of mitotic centrosome separation / mitotic G2/M transition checkpoint / peptidyl-threonine phosphorylation / inner cell mass cell proliferation / regulation of double-strand break repair via homologous recombination / nucleus organization ...negative regulation of mitotic nuclear division / apoptotic process involved in development / negative regulation of G0 to G1 transition / histone H3T11 kinase activity / regulation of mitotic centrosome separation / mitotic G2/M transition checkpoint / peptidyl-threonine phosphorylation / inner cell mass cell proliferation / regulation of double-strand break repair via homologous recombination / nucleus organization / negative regulation of gene expression, epigenetic / mitotic G2 DNA damage checkpoint signaling / Transcriptional Regulation by E2F6 / replicative senescence / Presynaptic phase of homologous DNA pairing and strand exchange / Activation of ATR in response to replication stress / Chk1/Chk2(Cds1) mediated inactivation of Cyclin B:Cdk1 complex / signal transduction in response to DNA damage / positive regulation of cell cycle / DNA damage checkpoint signaling / regulation of signal transduction by p53 class mediator / replication fork / condensed nuclear chromosome / TP53 Regulates Transcription of DNA Repair Genes / cellular response to mechanical stimulus / Signaling by SCF-KIT / G2/M DNA damage checkpoint / Ubiquitin-Mediated Degradation of Phosphorylated Cdc25A / G2/M transition of mitotic cell cycle / regulation of cell population proliferation / Processing of DNA double-strand break ends / Regulation of TP53 Activity through Phosphorylation / protein phosphorylation / protein kinase activity / DNA replication / non-specific serine/threonine protein kinase / chromatin remodeling / protein domain specific binding / protein serine kinase activity / DNA repair / protein serine/threonine kinase activity / apoptotic process / DNA damage response / centrosome / chromatin / protein-containing complex / : / nucleoplasm / ATP binding / nucleus / cytoplasm / cytosol Similarity search - Function
Checkpoint kinase 1, catalytic domain / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site ...Checkpoint kinase 1, catalytic domain / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha Beta Similarity search - Domain/homology
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
















 PDBj
PDBj











 Assembly
Assembly

 Spodoptera frugiperda (fall armyworm)
Spodoptera frugiperda (fall armyworm)
 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4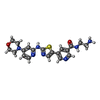
 Mass: 425.507 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H23N7O2S
Mass: 425.507 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H23N7O2S Mass: 18.015 Da / Num. of mol.: 222 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 222 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing