 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4fmu: Crystal structure of Methyltransferase domain of human SET domain... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4fmu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Methyltransferase domain of human SET domain-containing protein 2 Compound: Pr-SNF | ||||||
 Components Components | Histone-lysine N-methyltransferase SETD2 | ||||||
 Keywords Keywords | TRANSFERASE / Structural Genomics Consortium / SGC / Methyltransferase / SET domain-containing protein 2 / Pr-SNF | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptidyl-lysine trimethylation / microtubule cytoskeleton organization involved in mitosis / [histone H3]-lysine36 N-trimethyltransferase / regulation of mRNA export from nucleus / histone H3K36 trimethyltransferase activity / histone H3K36 methyltransferase activity / nucleosome organization / response to alkaloid / response to type I interferon / protein-lysine N-methyltransferase activity ...peptidyl-lysine trimethylation / microtubule cytoskeleton organization involved in mitosis / [histone H3]-lysine36 N-trimethyltransferase / regulation of mRNA export from nucleus / histone H3K36 trimethyltransferase activity / histone H3K36 methyltransferase activity / nucleosome organization / response to alkaloid / response to type I interferon / protein-lysine N-methyltransferase activity / positive regulation of ossification / regulation of protein localization to chromatin / response to metal ion / endodermal cell differentiation / histone H3 methyltransferase activity / regulation of double-strand break repair via homologous recombination / positive regulation of interferon-alpha production / mismatch repair / alpha-tubulin binding / positive regulation of autophagy / Transferases; Transferring one-carbon groups; Methyltransferases / stem cell differentiation / regulation of cytokinesis / transcription elongation by RNA polymerase II / PKMTs methylate histone lysines / chromosome / regulation of gene expression / defense response to virus / regulation of DNA-templated transcription / nucleoplasm / metal ion binding / nucleus / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Dong, A. / Zeng, H. / Ibanez, G. / Zheng, W. / Tempel, W. / Bountra, C. / Arrowsmith, C.H. / Edwards, A.M. / Brown, P.J. / Min, J. ...Dong, A. / Zeng, H. / Ibanez, G. / Zheng, W. / Tempel, W. / Bountra, C. / Arrowsmith, C.H. / Edwards, A.M. / Brown, P.J. / Min, J. / Luo, M. / Wu, H. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: J.Am.Chem.Soc. / Year: 2012 Title: Sinefungin Derivatives as Inhibitors and Structure Probes of Protein Lysine Methyltransferase SETD2. Authors: Zheng, W. / Ibanez, G. / Wu, H. / Blum, G. / Zeng, H. / Dong, A. / Li, F. / Hajian, T. / Allali-Hassani, A. / Amaya, M.F. / Siarheyeva, A. / Yu, W. / Brown, P.J. / Schapira, M. / Vedadi, M. / Min, J. / Luo, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4fmu.cif.gz | 66.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4fmu.ent.gz | 45.9 KB | Display | PDB format |
| PDBx/mmJSON format | 4fmu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fm/4fmuftp://data.pdbj.org/pub/pdb/validation_reports/fm/4fmu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4h12C  3h6l S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 31955.488 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: SETD2, HIF1, HYPB, KIAA1732, KMT3A, SET2, HSPC069 / Plasmid: pET28-MHL / Production host:  References: UniProt: Q9BYW2, histone-lysine N-methyltransferase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-0UM / ( | 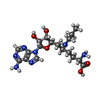  Mass: 423.467 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H29N7O5 Mass: 423.467 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H29N7O5#4: Chemical | ChemComp-UNX /   Num. of mol.: 13 / Source method: obtained synthetically Num. of mol.: 13 / Source method: obtained synthetically#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.46 Å3/Da / Density % sol: 49.95 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 20% PEG4000, 10% isoprpanol, 0.1M HEPES pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 23-ID-B / Wavelength: 1.03321 Å / Beamline: 23-ID-B / Wavelength: 1.03321 Å |
| Detector | Type: MAR 300 CCD / Detector: CCD / Date: Nov 11, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.03321 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→50 Å / Num. all: 19073 / Num. obs: 18501 / % possible obs: 97 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 7 % / Biso Wilson estimate: 38.63 Å2 / Rmerge(I) obs: 0.059 / Rsym value: 0.059 / Net I/σ(I): 42.7 |
| Reflection shell | Resolution: 2.1→2.14 Å / Redundancy: 6.4 % / Rmerge(I) obs: 0.221 / Mean I/σ(I) obs: 6.08 / Num. unique all: 769 / Rsym value: 0.221 / % possible all: 82.3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3H6L 3h6l Resolution: 2.1→35.12 Å / Cor.coef. Fo:Fc: 0.9135 / Cor.coef. Fo:Fc free: 0.8853 / SU R Cruickshank DPI: 0.174 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 46.23 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.262 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→35.12 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.23 Å / Total num. of bins used: 9
|
