- PDB-4f28: The Crystal Structure of a Human MitoNEET mutant with Met 62 Repl... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4f28
Title
The Crystal Structure of a Human MitoNEET mutant with Met 62 Replaced by a Gly
Components
CDGSH iron-sulfur domain-containing protein 1
Keywords
METAL BINDING PROTEIN / 2FE-2S PROTEINS / MEMBRANE / SIGNAL-ANCHOR / TRANSMEMBRANE METAL BINDING PROTEIN / PROTEIN FRUSTRATION / MITOCHONDRIAL OUTER MEMBRANE
Function / homology
Function and homology information
cysteine transaminase / L-cysteine:2-oxoglutarate transaminase activity / regulation of cellular respiration / regulation of autophagy / protein maturation / 2 iron, 2 sulfur cluster binding / pyridoxal phosphate binding / intracellular iron ion homeostasis / mitochondrial outer membrane / protein homodimerization activity ...cysteine transaminase / L-cysteine:2-oxoglutarate transaminase activity / regulation of cellular respiration / regulation of autophagy / protein maturation / 2 iron, 2 sulfur cluster binding / pyridoxal phosphate binding / intracellular iron ion homeostasis / mitochondrial outer membrane / protein homodimerization activity / mitochondrion / metal ion binding / identical protein binding Similarity search - Function
CDGSH iron-sulfur domain, mitoNEET-type / Iron sulphur domain-containing, mitoNEET, N-terminal / Iron-containing outer mitochondrial membrane protein N-terminus / CDGSH iron-sulfur domain-containing protein 1/2 / Iron-binding zinc finger CDGSH type / Iron-binding zinc finger, CDGSH type / MitoNEET, CDGSH iron-sulfur domain / CDGSH-type zinc finger. Function unknown. / Ribosomal Protein L9; domain 1 / 3-Layer(aba) Sandwich / Alpha Beta Similarity search - Domain/homology
Monochromator: Double crystal monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.9795 Å / Relative weight: 1
Reflection
Resolution: 1.55→33.87 Å / Num. obs: 20457 / % possible obs: 98.4 % / Observed criterion σ(I): -3 / Biso Wilson estimate: 33.164 Å2 / Rmerge(I) obs: 0.047 / Net I/σ(I): 25.26
Reflection shell
Diffraction-ID: 1
Resolution (Å)
Highest resolution (Å)
Rmerge(I) obs
Mean I/σ(I) obs
Num. measured obs
Num. unique obs
% possible all
1.55-1.61
0.991
1.56
11638
1899
87.2
1.61-1.67
0.703
3.53
26823
1926
99.8
1.67-1.75
0.412
5.82
32463
2113
100
1.75-1.84
0.266
8.02
30372
1976
99.5
1.84-1.95
0.174
12.3
30235
1982
99.8
1.95-2.1
0.108
19.22
31139
2039
100
2.1-2.31
0.082
27.68
31729
2073
99.9
2.31-2.65
0.056
41.05
32472
2122
100
2.65-3.33
0.039
57.65
31790
2104
100
3.33
0.034
67.28
31513
2223
98.2
-
Phasing
Phasing
Method: molecular replacement
Phasing MR
Rfactor: 43.72 / Model details: Phaser MODE: MR_AUTO
Highest resolution
Lowest resolution
Rotation
2.5 Å
33.87 Å
Translation
2.5 Å
33.87 Å
-
Processing
Software
Name
Version
Classification
NB
XSCALE
datascaling
PHASER
2.1.4
phasing
BUSTER-TNT
refinement
PDB_EXTRACT
3.11
dataextraction
Blu-Ice
datacollection
XDS
datareduction
BUSTER
2.8.0
refinement
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.55→33.87 Å / Cor.coef. Fo:Fc: 0.9484 / Cor.coef. Fo:Fc free: 0.9328 / Occupancy max: 1 / Occupancy min: 0.5 / Cross valid method: THROUGHOUT / σ(F): 0 Details: 1. ATOM RECORD CONTAINS SUM OF TLS AND RESIDUAL B FACTORS. ANISOU RECORD CONTAINS SUM OF TLS AND RESIDUAL U FACTORS. 2. NCS RESTRAINTS WERE APPLIED USING BUSTER'S LSSR RESTRAINT ...Details: 1. ATOM RECORD CONTAINS SUM OF TLS AND RESIDUAL B FACTORS. ANISOU RECORD CONTAINS SUM OF TLS AND RESIDUAL U FACTORS. 2. NCS RESTRAINTS WERE APPLIED USING BUSTER'S LSSR RESTRAINT REPRESENTATION (-AUTONCS). DIFFERENCE ELECTRON DENSITY, MOST LIKELY CORRESPONDING TO PROTEIN RESIDUES AT THE N-TERMINAL END OF CHAINS A OR B COULD NOT BE DEFINITIVELY ASSIGNED AND WERE NOT MODELED
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads






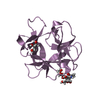








 PDBj
PDBj







 Assembly
Assembly

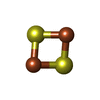
 Mass: 175.820 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe2S2
Mass: 175.820 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe2S2 Mass: 18.015 Da / Num. of mol.: 84 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 84 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL9-2 / Wavelength: 0.9795 Å
/ Beamline: BL9-2 / Wavelength: 0.9795 Å Processing
Processing