 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4eko: Initial Thaumatin Structure for Radiation Damage Experiment at 180 K -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4eko | ||||||
|---|---|---|---|---|---|---|---|
| Title | Initial Thaumatin Structure for Radiation Damage Experiment at 180 K | ||||||
 Components Components | Thaumatin-1 | ||||||
 Keywords Keywords | PLANT PROTEIN / sweet protein / radiation damage | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Thaumatococcus daniellii (katemfe) Thaumatococcus daniellii (katemfe) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.52 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.52 Å | ||||||
 Authors Authors | Warkentin, M. / Badeau, R. / Hopkins, J.B. / Thorne, R.E. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2012 Title: Spatial distribution of radiation damage to crystalline proteins at 25-300 K. Authors: Warkentin, M. / Badeau, R. / Hopkins, J.B. / Thorne, R.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4eko.cif.gz | 61 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4eko.ent.gz | 43.7 KB | Display | PDB format |
| PDBx/mmJSON format | 4eko.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ek/4ekoftp://data.pdbj.org/pub/pdb/validation_reports/ek/4eko | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4ek0C  4ekaC  4ekbC  4ekhC  4ektC  4el2C  4el3C  4el7C  4elaC  4ep8C  4epbC  4epdC  4epeC C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22243.119 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Thaumatococcus daniellii (katemfe) / References: UniProt: P02883 |
|---|---|
| #2: Chemical | ChemComp-TLA / 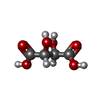  Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 337 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 337 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
| Sequence details | THAUMATIN WAS PURCHASED FROM SIGMA-ALDRICH (CATALOG # T7638) AND CONTAINED A MIXTURE OF THAUMATIN I ...THAUMATIN WAS PURCHASED FROM SIGMA-ALDRICH (CATALOG # T7638) AND CONTAINED A MIXTURE OF THAUMATIN I AND THAUMATIN II. LYS46 WAS USED IN PLACE OF ASN46 AND ASP113 WAS USED IN PLACE OF ASN113 AS WAS DONE IN ENTRY 1RQW AND AS SUGGESTED BY KO ET AL., ACTA CRYST. D50,813(1994). |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.81 Å3/Da / Density % sol: 56.26 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6.8 Details: sodium potassium tartrate, pH 6.8, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 180 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: CHESS  / Beamline: F1 / Wavelength: 0.917 Å / Beamline: F1 / Wavelength: 0.917 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 270 / Detector: CCD / Date: Jun 22, 2010 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: SI / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.917 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.52→99 Å / Num. obs: 39358 / % possible obs: 98.1 % / Redundancy: 5.7 % / Rmerge(I) obs: 0.075 / Χ2: 1.132 / Net I/σ(I): 8.8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.52→15.21 Å / Cor.coef. Fo:Fc: 0.9585 / Cor.coef. Fo:Fc free: 0.9533 / Occupancy max: 1 / Occupancy min: 0.11 / SU R Cruickshank DPI: 0.064 / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 104.45 Å2 / Biso mean: 19.1655 Å2 / Biso min: 7.53 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.151 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.52→15.21 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.52→1.56 Å / Total num. of bins used: 20
|
