 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4czg | ||||||
|---|---|---|---|---|---|---|---|
| Title | C. crescentus MreB, single filament, ADP, A22 inhibitor | ||||||
 Components Components | ROD SHAPE-DETERMINING PROTEIN MREB | ||||||
 Keywords Keywords | STRUCTURAL PROTEIN / BACTERIAL ACTIN / BACTERIAL CYTOSKELETON | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  CAULOBACTER VIBRIOIDES (bacteria) CAULOBACTER VIBRIOIDES (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | Lowe, J. / van den Ent, F. | ||||||
 Citation Citation | Journal: Elife / Year: 2014 Title: Bacterial Actin Mreb Forms Antiparallel Double Filaments. Authors: Van Den Ent, F. / Izore, T. / Bharat, T.A. / Johnson, C.M. / Lowe, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4czg.cif.gz | 201.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4czg.ent.gz | 160.3 KB | Display | PDB format |
| PDBx/mmJSON format | 4czg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cz/4czgftp://data.pdbj.org/pub/pdb/validation_reports/cz/4czg | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4czeC  4czfC  4czhC  4cziC  4czjC  4czkC  4czlC  4czmC  1jceS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 36865.496 Da / Num. of mol.: 1 / Fragment: RESIDUES 9-347 / Mutation: YES Source method: isolated from a genetically manipulated source Details: M-I9-CCMREB(F102S, V103G)-A347-GSHHHHHH / Source: (gene. exp.) CAULOBACTER VIBRIOIDES (bacteria) / Plasmid: PHIS17 / Production host: |
|---|---|
| #2: Chemical | ChemComp-QH3 / 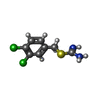  Mass: 235.134 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H8Cl2N2S / Comment: antibiotic*YM Mass: 235.134 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H8Cl2N2S / Comment: antibiotic*YM |
| #3: Chemical | ChemComp-ADP /   Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM |
| #4: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 434 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 434 / Source method: isolated from a natural source / Formula: H2O |
| Nonpolymer details | A22, S-(3,4-DICHLOROBE |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.68 Å3/Da / Density % sol: 54.14 % / Description: NONE |
|---|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU FR-E / Wavelength: 1.5418 |
| Detector | Type: MARESEARCH / Detector: IMAGE PLATE |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→50 Å / Num. obs: 52173 / % possible obs: 87.8 % / Observed criterion σ(I): 0 / Redundancy: 6.8 % / Biso Wilson estimate: 20.8 Å2 / Rmerge(I) obs: 0.05 / Net I/σ(I): 19.8 |
| Reflection shell | Resolution: 1.5→1.58 Å / Redundancy: 5.4 % / Rmerge(I) obs: 0.69 / Mean I/σ(I) obs: 1.1 / % possible all: 77 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1JCE Resolution: 1.5→20.397 Å / SU ML: 0.45 / σ(F): 1.34 / Phase error: 23.25 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.77 Å / VDW probe radii: 0.9 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 60.873 Å2 / ksol: 0.459 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→20.397 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
