SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AB" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "BA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "CC" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL. THE SHEETS PRESENTED AS "DA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 8-STRANDED BARREL THIS IS REPRESENTED BY A 9-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.979 Å / Relative weight: 1
Reflection
Resolution: 1.39→78.43 Å / Num. obs: 408156 / % possible obs: 99.9 % / Observed criterion σ(I): 2 / Redundancy: 6.7 % / Rmerge(I) obs: 0.08 / Net I/σ(I): 12.5
Reflection shell
Resolution: 1.39→1.43 Å / Redundancy: 6.7 % / Rmerge(I) obs: 0.62 / Mean I/σ(I) obs: 2.7 / % possible all: 100
-
Processing
Software
Name
Version
Classification
PHENIX
(PHENIX.REFINE: 1.8_1069)
refinement
XDS
datareduction
XSCALE
datascaling
PHASER
phasing
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.39→67.334 Å / SU ML: 0.11 / σ(F): 1.93 / Phase error: 14.05 / Stereochemistry target values: ML Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. NITRIC OXIDE LIGAND WAS REFINED IN REAL SPACE WITH COOT
Rfactor
Num. reflection
% reflection
Rfree
0.1571
20519
5.03 %
Rwork
0.1302
-
-
obs
0.1315
408086
99.9 %
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL
Refinement step
Cycle: LAST / Resolution: 1.39→67.334 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
16566
0
430
1713
18709
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.006
17708
X-RAY DIFFRACTION
f_angle_d
1.197
24191
X-RAY DIFFRACTION
f_dihedral_angle_d
17.369
6642
X-RAY DIFFRACTION
f_chiral_restr
0.08
2384
X-RAY DIFFRACTION
f_plane_restr
0.006
3241
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
1.39-1.4058
0.222
669
0.1715
12876
X-RAY DIFFRACTION
100
1.4058-1.4223
0.2166
692
0.1608
12896
X-RAY DIFFRACTION
100
1.4223-1.4397
0.2111
661
0.1553
12923
X-RAY DIFFRACTION
100
1.4397-1.4579
0.1891
655
0.1472
12983
X-RAY DIFFRACTION
100
1.4579-1.4771
0.2009
657
0.1426
12872
X-RAY DIFFRACTION
100
1.4771-1.4973
0.1845
701
0.1373
12864
X-RAY DIFFRACTION
100
1.4973-1.5187
0.188
625
0.1329
12936
X-RAY DIFFRACTION
100
1.5187-1.5414
0.1774
693
0.1288
12900
X-RAY DIFFRACTION
100
1.5414-1.5655
0.1702
688
0.1245
12941
X-RAY DIFFRACTION
100
1.5655-1.5912
0.1607
683
0.1135
12911
X-RAY DIFFRACTION
100
1.5912-1.6186
0.1547
680
0.114
12922
X-RAY DIFFRACTION
100
1.6186-1.648
0.1689
668
0.1135
12907
X-RAY DIFFRACTION
100
1.648-1.6797
0.1606
662
0.1132
12927
X-RAY DIFFRACTION
100
1.6797-1.714
0.1533
667
0.1135
12872
X-RAY DIFFRACTION
100
1.714-1.7513
0.1614
689
0.1152
12942
X-RAY DIFFRACTION
100
1.7513-1.792
0.156
689
0.1149
12868
X-RAY DIFFRACTION
100
1.792-1.8368
0.153
684
0.1135
12919
X-RAY DIFFRACTION
100
1.8368-1.8865
0.1569
688
0.1203
12935
X-RAY DIFFRACTION
100
1.8865-1.942
0.1635
696
0.1227
12884
X-RAY DIFFRACTION
100
1.942-2.0047
0.1582
730
0.1231
12911
X-RAY DIFFRACTION
100
2.0047-2.0764
0.153
686
0.127
12954
X-RAY DIFFRACTION
100
2.0764-2.1595
0.1597
670
0.1235
12910
X-RAY DIFFRACTION
100
2.1595-2.2578
0.153
691
0.1245
12925
X-RAY DIFFRACTION
100
2.2578-2.3768
0.1528
655
0.1294
12943
X-RAY DIFFRACTION
100
2.3768-2.5258
0.1505
745
0.1316
12894
X-RAY DIFFRACTION
100
2.5258-2.7208
0.1466
697
0.1359
12933
X-RAY DIFFRACTION
100
2.7208-2.9946
0.1613
724
0.1441
12913
X-RAY DIFFRACTION
100
2.9946-3.4279
0.1565
693
0.1481
12906
X-RAY DIFFRACTION
100
3.4279-4.3187
0.146
681
0.1278
12981
X-RAY DIFFRACTION
100
4.3187-67.4131
0.134
700
0.131
13019
X-RAY DIFFRACTION
99
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information CORYNEBACTERIUM GLUTAMICUM ATCC 13032 (bacteria)
CORYNEBACTERIUM GLUTAMICUM ATCC 13032 (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj




 Assembly
Assembly

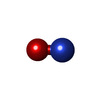

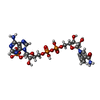


 Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4
Mass: 616.487 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 30.006 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: NO
Mass: 30.006 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: NO Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 745.421 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H30N7O17P3
Mass: 745.421 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 209.240 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H19NO5 / Comment: pH buffer*YM
Mass: 209.240 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H19NO5 / Comment: pH buffer*YM Sample preparation
Sample preparation / Beamline: I02 / Wavelength: 0.979
/ Beamline: I02 / Wavelength: 0.979  Processing
Processing