 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4b5p: Crystal structure of human alpha tubulin acetyltransferase cataly... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4b5p | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human alpha tubulin acetyltransferase catalytic domain Q58A variant | ||||||
 Components Components | (ALPHA-TUBULIN N-ACETYLTRANSFERASE) x 2 | ||||||
 Keywords Keywords | TRANSFERASE / LYSINE ACETYLTRANSFERASE / ACETYL COA / TUBULIN / MICROTUBULES / CILIUM / INTRAFLAGELLAR TRANSPORT | ||||||
| Function / homology |  Function and homology information Function and homology informationalpha-tubulin N-acetyltransferase / tubulin N-acetyltransferase activity / alpha-tubulin acetylation / microtubule bundle / Cargo trafficking to the periciliary membrane / L-lysine N6-acetyltransferase activity, acting on acetyl phosphate as donor / response to pain / neuron development / response to mechanical stimulus / cilium assembly ...alpha-tubulin N-acetyltransferase / tubulin N-acetyltransferase activity / alpha-tubulin acetylation / microtubule bundle / Cargo trafficking to the periciliary membrane / L-lysine N6-acetyltransferase activity, acting on acetyl phosphate as donor / response to pain / neuron development / response to mechanical stimulus / cilium assembly / clathrin-coated pit / regulation of microtubule cytoskeleton organization / microtubule cytoskeleton organization / mitotic spindle / microtubule / axon / focal adhesion / cytosol Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Taschner, M. / Vetter, M. / Lorentzen, E. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2012 Title: Atomic Resolution Structure of Human Alpha-Tubulin Acetyltransferase Bound to Acetyl-Coa. Authors: Taschner, M. / Vetter, M. / Lorentzen, E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4b5p.cif.gz | 235.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4b5p.ent.gz | 194.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4b5p.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/b5/4b5pftp://data.pdbj.org/pub/pdb/validation_reports/b5/4b5p | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4b5oSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22785.117 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN, RESIDUES 1-196 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  References: UniProt: Q5SQI0, alpha-tubulin N-acetyltransferase | ||||
|---|---|---|---|---|---|
| #2: Protein | Mass: 22799.143 Da / Num. of mol.: 1 / Fragment: CATALYTIC DOMAIN, RESIDUES 1-196 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host: References: UniProt: Q5SQI0, alpha-tubulin N-acetyltransferase | ||||
| #3: Chemical |   Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Na#4: Chemical | 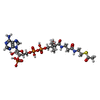  Mass: 809.571 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C23H38N7O17P3S Mass: 809.571 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C23H38N7O17P3S#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 350 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 350 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.54 Å3/Da / Density % sol: 51.57 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7 / Details: 2.8 M NA ACETATE PH 7.0 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06DA / Wavelength: 0.9999 / Beamline: X06DA / Wavelength: 0.9999 |
| Detector | Type: DECTRIS PILATUS / Detector: PIXEL / Date: Apr 12, 2012 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9999 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→40 Å / Num. obs: 60072 / % possible obs: 1 % / Observed criterion σ(I): -2 / Redundancy: 6.6 % / Biso Wilson estimate: 25.1 Å2 / Rmerge(I) obs: 0.04 / Net I/σ(I): 24.7 |
| Reflection shell | Resolution: 1.6→1.7 Å / Redundancy: 6.3 % / Rmerge(I) obs: 0.74 / Mean I/σ(I) obs: 2.7 / % possible all: 0.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4B5O Resolution: 1.6→39.669 Å / SU ML: 0.24 / σ(F): 1.99 / Phase error: 22.55 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.98 Å / VDW probe radii: 1.2 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 59.684 Å2 / ksol: 0.4 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→39.669 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
