 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4avk: Structure of trigonal FimH lectin domain crystal soaked with an a... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4avk | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of trigonal FimH lectin domain crystal soaked with an alpha- D-mannoside O-linked to propynyl pyridine at 2.4A resolution | ||||||
 Components Components | FIMH | ||||||
 Keywords Keywords | CELL ADHESION / BACTERIAL ADHESIN / TYPE 1 FIMBRIAE / URINARY TRACT INFECTION / VARIABLE IMMUNOGLOBULIN FOLD | ||||||
| Function / homology |  Function and homology information Function and homology informationpilus tip / mechanosensory behavior / Attachment of bacteria to epithelial cells / cell adhesion involved in single-species biofilm formation / pilus / cell-substrate adhesion / D-mannose binding / host cell membrane / cell adhesion Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Wellens, A. / Lahmann, M. / Touaibia, M. / Vaucher, J. / Oscarson, S. / Roy, R. / Remaut, H. / Bouckaert, J. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2012 Title: The Tyrosine Gate as a Potential Entropic Lever in the Receptor-Binding Site of the Bacterial Adhesin Fimh. Authors: Wellens, A. / Lahmann, M. / Touaibia, M. / Vaucher, J. / Oscarson, S. / Roy, R. / Remaut, H. / Bouckaert, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4avk.cif.gz | 78.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4avk.ent.gz | 59.4 KB | Display | PDB format |
| PDBx/mmJSON format | 4avk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/av/4avkftp://data.pdbj.org/pub/pdb/validation_reports/av/4avk | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4auuC  4auyC  4av0C  4av4C  4av5C  4avhC  4aviC  4avjC  2vcoS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |





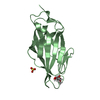

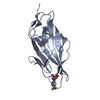




-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 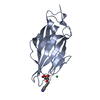
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 16916.828 Da / Num. of mol.: 2 / Fragment: LECTIN DOMAIN, RESIDUES 10-167 Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Sugar | 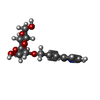  Type: D-saccharide / Mass: 295.288 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H17NO6 Type: D-saccharide / Mass: 295.288 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H17NO6#3: Chemical |   Mass: 58.693 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ni Mass: 58.693 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ni#4: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 171 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 171 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.76 Å3/Da / Density % sol: 55.51 % / Description: NONE |
|---|---|
| Crystal grow | pH: 8.6 Details: 1 M LI2SO4, 100 MM TRIS PH 8.6, 10 MM NICL2, 0.2 M NON-DETERGENT SULFOBETAINE 201 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-2 / Wavelength: 1.016 / Beamline: ID23-2 / Wavelength: 1.016 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Nov 23, 2010 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.016 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→25 Å / Num. obs: 16232 / % possible obs: 99.9 % / Observed criterion σ(I): 0 / Redundancy: 7.2 % / Biso Wilson estimate: 27.9 Å2 / Rmerge(I) obs: 0.11 / Net I/σ(I): 14 |
| Reflection shell | Resolution: 2.4→2.6 Å / Redundancy: 6.7 % / Rmerge(I) obs: 0.38 / Mean I/σ(I) obs: 5.2 / % possible all: 92.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2VCO Resolution: 2.4→24.811 Å / SU ML: 0.26 / σ(F): 2.03 / Phase error: 19.72 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.73 Å / VDW probe radii: 1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 26.133 Å2 / ksol: 0.351 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→24.811 Å
| ||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
