 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4asq | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of ANCE in complex with Bradykinin | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | HYDROLASE / ZINC METALLOPROTEASE / SUBSTRATE RECOGNITION | |||||||||
| Function / homology |  Function and homology information Function and homology informationMetabolism of Angiotensinogen to Angiotensins / metamorphosis / mating / response to symbiotic bacterium / peptidyl-dipeptidase A / negative regulation of cell adhesion / peptide hormone processing / negative regulation of blood coagulation / peptidyl-dipeptidase activity / cysteine-type endopeptidase inhibitor activity ...Metabolism of Angiotensinogen to Angiotensins / metamorphosis / mating / response to symbiotic bacterium / peptidyl-dipeptidase A / negative regulation of cell adhesion / peptide hormone processing / negative regulation of blood coagulation / peptidyl-dipeptidase activity / cysteine-type endopeptidase inhibitor activity / carboxypeptidase activity / : / negative regulation of proteolysis / platelet alpha granule lumen / Peptide ligand-binding receptors / Post-translational protein phosphorylation / hormone activity / vasodilation / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / metallopeptidase activity / blood coagulation / Platelet degranulation / heparin binding / extracellular matrix / positive regulation of cytosolic calcium ion concentration / blood microparticle / G alpha (i) signalling events / G alpha (q) signalling events / positive regulation of apoptotic process / endoplasmic reticulum lumen / inflammatory response / signaling receptor binding / proteolysis / : / extracellular exosome / extracellular region / zinc ion binding / metal ion binding / plasma membrane Similarity search - Function | |||||||||
| Biological species |   HOMO SAPIENS (human) HOMO SAPIENS (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.99 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.99 Å | |||||||||
 Authors Authors | Akif, M. / Masuyer, G. / Sturrock, E.D. / Isaac, R.E. / Acharya, K.R. | |||||||||
 Citation Citation | Journal: FEBS J. / Year: 2012 Title: Structural Basis of Peptide Recognition by the Angiotensin-I Converting Enzyme Homologue Ance from Drosophila Melanogaster Authors: Akif, M. / Masuyer, G. / Bingham, R.J. / Sturrock, E.D. / Isaac, R.E. / Acharya, K.R. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4asq.cif.gz | 274.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4asq.ent.gz | 221.2 KB | Display | PDB format |
| PDBx/mmJSON format | 4asq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/as/4asqftp://data.pdbj.org/pub/pdb/validation_reports/as/4asq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4aa1C  4aa2C  4asrC  2x8yS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
























-Links
 PDBj
PDBj














- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein / Protein/peptide , 2 types, 2 molecules AP
| #1: Protein | Mass: 69152.602 Da / Num. of mol.: 1 / Fragment: RESIDUES 17-614 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  KOMAGATAELLA PASTORIS (fungus) / Strain (production host): GS115 / References: UniProt: Q10714, peptidyl-dipeptidase A KOMAGATAELLA PASTORIS (fungus) / Strain (production host): GS115 / References: UniProt: Q10714, peptidyl-dipeptidase A |
|---|---|
| #2: Protein/peptide | Mass: 1062.224 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) HOMO SAPIENS (human) / References: UniProt: P01042 |
-Sugars , 2 types, 3 molecules 
| #3: Polysaccharide | beta-D-mannopyranose-(1-6)-alpha-D-mannopyranose-(1-3)-[alpha-D-mannopyranose-(1-6)]beta-D- ...beta-D-mannopyranose-(1-6)-alpha-D-mannopyranose-(1-3)-[alpha-D-mannopyranose-(1-6)]beta-D-mannopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source |
|---|---|
| #5: Sugar |  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2Source method: isolated from a genetically manipulated source Formula: C8H15NO6 |
-Non-polymers , 3 types, 502 molecules 

| #4: Chemical | ChemComp-ZN /  Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
|---|---|
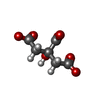
| #6: Chemical | ChemComp-FLC /  Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 |
| #7: Water | ChemComp-HOH / Mass: 18.015 Da / Num. of mol.: 500 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.2 Å3/Da / Density % sol: 71 % / Description: NONE |
|---|---|
| Crystal grow | pH: 7.5 / Details: 100 MM HEPES 1.3 M SODIUM CITRATE, pH 7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I03 / Wavelength: 0.9763 / Beamline: I03 / Wavelength: 0.9763 |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Dec 9, 2009 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9763 Å / Relative weight: 1 |
| Reflection | Resolution: 1.97→50 Å / Num. obs: 89919 / % possible obs: 96 % / Observed criterion σ(I): 0 / Redundancy: 2.3 % / Rmerge(I) obs: 0.09 / Net I/σ(I): 9.2 |
| Reflection shell | Resolution: 1.9→1.97 Å / Redundancy: 1.8 % / Rmerge(I) obs: 0.42 / Mean I/σ(I) obs: 2.3 / % possible all: 83.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2X8Y Resolution: 1.99→86.56 Å / Cor.coef. Fo:Fc: 0.956 / Cor.coef. Fo:Fc free: 0.947 / SU B: 5.865 / SU ML: 0.077 / Cross valid method: THROUGHOUT / ESU R: 0.121 / ESU R Free: 0.112 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. ONLY RESIDUES 1-3 OF BRADYKININ ARE VISIBLE
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.318 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.99→86.56 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|