 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-4a0e: Crystal structure of the cytoplasmic N-terminal domain of Yersini... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4a0e | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the cytoplasmic N-terminal domain of Yersinia pestis YscD | ||||||
 Components Components | TYPE III SECRETION PROTEIN | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / SAD PHASING / TYPE III SECRETION SYSTEM | ||||||
| Function / homology |  Function and homology information Function and homology information: / Uncharacterised protein Y4YQ C-terminal domain / Yop protein translocation protein D, periplasmic domain / : / : / YscD/CdsD-like Bon-like domain 1 / YscD/CdsD-like Bon-like domain 3 / YscD/CdsD-like Bon-like domain 2 / Type III secretion system YscD/HrpQ / YscD, cytoplasmic domain ...: / Uncharacterised protein Y4YQ C-terminal domain / Yop protein translocation protein D, periplasmic domain / : / : / YscD/CdsD-like Bon-like domain 1 / YscD/CdsD-like Bon-like domain 3 / YscD/CdsD-like Bon-like domain 2 / Type III secretion system YscD/HrpQ / YscD, cytoplasmic domain / Inner membrane component of T3SS, cytoplasmic domain / Tumour Suppressor Smad4 - #20 / Tumour Suppressor Smad4 / Forkhead-associated (FHA) domain / SMAD/FHA domain superfamily / Sandwich / Mainly Beta Similarity search - Domain/homology | ||||||
| Biological species |   YERSINIA PESTIS (bacteria) YERSINIA PESTIS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.042 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 2.042 Å | ||||||
 Authors Authors | Lountos, G.T. / Tropea, J.E. / Waugh, D.S. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2012 Title: Structure of the Cytoplasmic Domain of Yersinia Pestis Yscd, an Essential Component of the Type III Secretion System Authors: Lountos, G.T. / Tropea, J.E. / Waugh, D.S. | ||||||
| History |
|
- Structure visualization
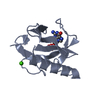
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4a0e.cif.gz | 96.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4a0e.ent.gz | 75.5 KB | Display | PDB format |
| PDBx/mmJSON format | 4a0e.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a0/4a0eftp://data.pdbj.org/pub/pdb/validation_reports/a0/4a0e | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 13483.413 Da / Num. of mol.: 2 / Fragment: CYTOPLASMIC DOMAIN, RESIDUES 2-121 Source method: isolated from a genetically manipulated source Source: (gene. exp.) YERSINIA PESTIS (bacteria) / Plasmid: PJT173 / Production host: #2: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 88 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 88 / Source method: isolated from a natural source / Formula: H2OSequence details | FIRST THREE GLYCINES ARE NON-NATIVE RESIDUES REMAINING AFTER TAG CLEAVAGE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.56 Å3/Da / Density % sol: 51.9 % / Description: NONE |
|---|---|
| Crystal grow | pH: 8.5 Details: 0.085M TRIS HCL PH 8.5, 25.5% (W/V) PEG 4000, 0.17M NA ACETATE, 15% (V/V) GLYCEROL, 10 MM TCEP HYDROCHLORIDE |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 22-ID / Wavelength: 1 / Beamline: 22-ID / Wavelength: 1 |
| Detector | Type: MARRESEARCH MX-300 / Detector: CCD / Date: Jun 6, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.04→50 Å / Num. obs: 18490 / % possible obs: 98.9 % / Observed criterion σ(I): 2.1 / Redundancy: 5.1 % / Biso Wilson estimate: 46.43 Å2 / Rmerge(I) obs: 0.06 / Net I/σ(I): 41.6 |
| Reflection shell | Resolution: 2.04→2.08 Å / Redundancy: 4 % / Rmerge(I) obs: 0.64 / Mean I/σ(I) obs: 2.1 / % possible all: 99.3 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD Starting model: NONE Resolution: 2.042→39.409 Å / SU ML: 0.4 / σ(F): 0 / Phase error: 23.14 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 1.06 Å / VDW probe radii: 1.3 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 48.686 Å2 / ksol: 0.352 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.042→39.409 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 28.8948 Å / Origin y: -14.1843 Å / Origin z: -46.6411 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: ALL |
