 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3zyc | ||||||
|---|---|---|---|---|---|---|---|
| Title | DYNAMIN 1 GTPASE GED FUSION DIMER COMPLEXED WITH GMPPCP | ||||||
 Components Components | DYNAMIN-1 | ||||||
 Keywords Keywords | HYDROLASE / MEMBRANE FISSION / NUCLEOTIDE-BINDING / ENDOCYTOSIS / MOTOR PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationclathrin coat assembly involved in endocytosis / vesicle scission / synaptic vesicle budding from presynaptic endocytic zone membrane / dynamin GTPase / chromaffin granule / regulation of vesicle size / Retrograde neurotrophin signalling / Toll Like Receptor 4 (TLR4) Cascade / Formation of annular gap junctions / endosome organization ...clathrin coat assembly involved in endocytosis / vesicle scission / synaptic vesicle budding from presynaptic endocytic zone membrane / dynamin GTPase / chromaffin granule / regulation of vesicle size / Retrograde neurotrophin signalling / Toll Like Receptor 4 (TLR4) Cascade / Formation of annular gap junctions / endosome organization / Gap junction degradation / Recycling pathway of L1 / phosphatidylinositol-3,4,5-trisphosphate binding / EPH-ephrin mediated repulsion of cells / endocytic vesicle / phosphatidylinositol-4,5-bisphosphate binding / clathrin-coated pit / MHC class II antigen presentation / receptor-mediated endocytosis / protein homooligomerization / endocytosis / GDP binding / Clathrin-mediated endocytosis / presynapse / microtubule binding / protein homotetramerization / microtubule / GTPase activity / synapse / protein kinase binding / GTP binding / protein homodimerization activity / RNA binding / extracellular exosome / identical protein binding / plasma membrane / cytoplasm Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Chappie, J.S. / Mears, J.A. / Fang, S. / Leonard, M. / Schmid, S.L. / Milligan, R.A. / Hinshaw, J.E. / Dyda, F. | ||||||
 Citation Citation | Journal: Cell / Year: 2011 Title: A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Authors: Joshua S Chappie / Jason A Mears / Shunming Fang / Marilyn Leonard / Sandra L Schmid / Ronald A Milligan / Jenny E Hinshaw / Fred Dyda /  Abstract: The GTPase dynamin catalyzes membrane fission by forming a collar around the necks of clathrin-coated pits, but the specific structural interactions and conformational changes that drive this process ...The GTPase dynamin catalyzes membrane fission by forming a collar around the necks of clathrin-coated pits, but the specific structural interactions and conformational changes that drive this process remain a mystery. We present the GMPPCP-bound structures of the truncated human dynamin 1 helical polymer at 12.2 Å and a fusion protein, GG, linking human dynamin 1's catalytic G domain to its GTPase effector domain (GED) at 2.2 Å. The structures reveal the position and connectivity of dynamin fragments in the assembled structure, showing that G domain dimers only form between tetramers in sequential rungs of the dynamin helix. Using chemical crosslinking, we demonstrate that dynamin tetramers are made of two dimers, in which the G domain of one molecule interacts in trans with the GED of another. Structural comparison of GG(GMPPCP) to the GG transition-state complex identifies a hydrolysis-dependent powerstroke that may play a role in membrane-remodeling events necessary for fission. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3zyc.cif.gz | 154.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3zyc.ent.gz | 119.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3zyc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zy/3zycftp://data.pdbj.org/pub/pdb/validation_reports/zy/3zyc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1949C  3zysC  2x2eS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj














- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-1, 0.0003, -0.0042), Vector: |
-Components
| #1: Protein | Mass: 39388.961 Da / Num. of mol.: 2 Fragment: GTPASE DOMAIN, RESIDUES 6-320, GTPASE EFFECTOR DOMAIN, RESIDUES 726-750 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  #2: Chemical | 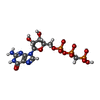  Mass: 521.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H18N5O13P3 / Comment: GMP-PCP, energy-carrying molecule analogue*YM Mass: 521.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H18N5O13P3 / Comment: GMP-PCP, energy-carrying molecule analogue*YM#3: Chemical |   Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 413 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 413 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.19 Å3/Da / Density % sol: 43.71 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7 Details: GGGMPPCP WAS CRYSTALLIZED BY SITTING DROP VAPOR DIFFUSION IN 0.1M HEPES PH 7.0, 200 MM MGCL2, AND 24-30% PEG 2000 MME USING A DROP SIZE OF 3 MICROL AND RESERVOIR VOLUME OF 65 MICROL. ...Details: GGGMPPCP WAS CRYSTALLIZED BY SITTING DROP VAPOR DIFFUSION IN 0.1M HEPES PH 7.0, 200 MM MGCL2, AND 24-30% PEG 2000 MME USING A DROP SIZE OF 3 MICROL AND RESERVOIR VOLUME OF 65 MICROL. CRYSTALS GREW IN 3-5 DAYS AT 20C. THE ADDITION OF 200 MM LICL, NACL, KCL, OR NH4CL TO THE RESERVOIR SOLUTION DRAMATICALLY IMPROVED THE CRYSTALS. CRYSTALS GROWN IN THE PRESENCE OF KCL SHOWED THE BEST DIFFRACTION. |
-Data collection
| Diffraction | Mean temperature: 95 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 HF / Wavelength: 1.5418 |
| Detector | Type: RIGAKU-MSC SATURN 200 / Detector: CCD / Date: Sep 15, 2010 / Details: CONFOCAL |
| Radiation | Monochromator: MULTILAYER MIRROR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→30 Å / Num. obs: 34206 / % possible obs: 99 % / Observed criterion σ(I): -3 / Redundancy: 4.27 % / Rmerge(I) obs: 0.08 / Net I/σ(I): 12.23 |
| Reflection shell | Resolution: 2.2→2.26 Å / Redundancy: 2.57 % / Rmerge(I) obs: 0.34 / Mean I/σ(I) obs: 2.95 / % possible all: 90.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2X2E Resolution: 2.2→30 Å / Rfactor Rfree error: 0.009 / Data cutoff high absF: 10000 / Data cutoff low absF: 0 / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 18.5092 Å2 / ksol: 0.34 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→30 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
