Mass: 18.015 Da / Num. of mol.: 472 / Source method: isolated from a natural source / Formula: H2O
Nonpolymer details
L-TRYPTOPHAN (LTR): THE BOUND L-TRYPTOPHAN ACTIVATES TRAP TO BIND MRNA TRANSCRIPT
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.55 Å3/Da / Density % sol: 51.7 % / Description: NONE
Crystal grow
Temperature: 293 K / pH: 7.5 Details: PROTEIN WAS IN BUFFER 50 MM OF POTASSIUM PHOSPHATE; CONCENTRATION 14.7 MG/ML, TEMPERATURE 293K; NO L-TRP WAS ADDED. CRYSTALLISATION CONDITIONS: 0.1 M HEPES PH 7.0, 5% (V/V) TASCIMATE PH 7.0, ...Details: PROTEIN WAS IN BUFFER 50 MM OF POTASSIUM PHOSPHATE; CONCENTRATION 14.7 MG/ML, TEMPERATURE 293K; NO L-TRP WAS ADDED. CRYSTALLISATION CONDITIONS: 0.1 M HEPES PH 7.0, 5% (V/V) TASCIMATE PH 7.0, 10% (W/V) PEG5KMME. CRYOPROTECTION: 15%(W/V) PEG5KMME, 10%(V/V) GLYCEROL.
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj
 Assembly
Assembly


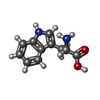
 Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 22 / Source method: obtained synthetically / Formula: C11H12N2O2
Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 22 / Source method: obtained synthetically / Formula: C11H12N2O2 Mass: 18.015 Da / Num. of mol.: 472 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 472 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: PX14.2 / Wavelength: 0.98
/ Beamline: PX14.2 / Wavelength: 0.98  Processing
Processing