 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
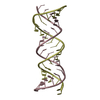
Yorodumi- PDB-3uun: Crystal Structure of N-terminal first spectrin repeat of dystrophin -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3uun | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of N-terminal first spectrin repeat of dystrophin | ||||||
 Components Components | Dystrophin | ||||||
 Keywords Keywords | STRUCTURAL PROTEIN / triple helical / Cell structure and stability / Cytoskeletal | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of muscle system process / regulation of cellular response to growth factor stimulus / syntrophin complex / cardiac muscle cell action potential / synaptic signaling / dystrophin-associated glycoprotein complex / cell-substrate junction / motile cilium assembly / peptide biosynthetic process / dystroglycan binding ...regulation of muscle system process / regulation of cellular response to growth factor stimulus / syntrophin complex / cardiac muscle cell action potential / synaptic signaling / dystrophin-associated glycoprotein complex / cell-substrate junction / motile cilium assembly / peptide biosynthetic process / dystroglycan binding / regulation of skeletal muscle contraction by regulation of release of sequestered calcium ion / vinculin binding / camera-type eye development / regulation of sodium ion transmembrane transport / costamere / Formation of the dystrophin-glycoprotein complex (DGC) / muscle cell development / regulation of calcium ion transmembrane transport / Striated Muscle Contraction / muscle cell cellular homeostasis / filopodium membrane / muscle organ development / structural constituent of muscle / maintenance of blood-brain barrier / myosin binding / neuron projection terminus / nitric-oxide synthase binding / regulation of skeletal muscle contraction / skeletal muscle tissue development / Non-integrin membrane-ECM interactions / neuron development / response to muscle stretch / cardiac muscle contraction / regulation of release of sequestered calcium ion into cytosol by sarcoplasmic reticulum / regulation of cardiac muscle contraction by regulation of the release of sequestered calcium ion / positive regulation of neuron differentiation / regulation of heart rate / filopodium / positive regulation of neuron projection development / sarcolemma / Z disc / structural constituent of cytoskeleton / intracellular protein localization / actin binding / protein-containing complex assembly / cytoskeleton / postsynaptic membrane / membrane raft / synapse / cell surface / protein-containing complex / zinc ion binding / nucleus / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Muthu, M. / Richardson, K.A. / Sutherland-smith, A.J. | ||||||
 Citation Citation | Journal: Plos One / Year: 2012 Title: The crystal structures of dystrophin and utrophin spectrin repeats: implications for domain boundaries Authors: Muthu, M. / Richardson, K.A. / Sutherland-Smith, A.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3uun.cif.gz | 103.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3uun.ent.gz | 81.7 KB | Display | PDB format |
| PDBx/mmJSON format | 3uun.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/uu/3uunftp://data.pdbj.org/pub/pdb/validation_reports/uu/3uun | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components on special symmetry positions |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Ens-ID: 1 / Beg auth comp-ID: LEU / Beg label comp-ID: LEU / Refine code: 4
NCS oper:
|
-Components
| #1: Protein | Mass: 13642.204 Da / Num. of mol.: 2 / Fragment: Spectrin Repeat, UNP residues 338-456 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: DMD / Plasmid: pPRoExHTb / Production host:  #2: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 34 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 34 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.03 Å3/Da / Density % sol: 39.52 % |
|---|---|
| Crystal grow | Temperature: 294.15 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: 0.1M Tris HCl, 2.0M Ammonium sulphate, pH 8.5, VAPOR DIFFUSION, HANGING DROP, temperature 294.15K |
-Data collection
| Diffraction | Mean temperature: 120 K | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 / Wavelength: 1.5418 Å | |||||||||||||||
| Detector | Type: RIGAKU RAXIS IV++ / Detector: IMAGE PLATE / Date: Apr 18, 2010 / Details: OSMIC BLUE CONFOCAL MIRRORS | |||||||||||||||
| Radiation | Monochromator: OSMIC MIRRORS / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 | |||||||||||||||
| Reflection twin |
| |||||||||||||||
| Reflection | Resolution: 2.3→33.24 Å / Num. all: 9999 / Num. obs: 9035 / % possible obs: 99.9 % / Observed criterion σ(F): 3.1 / Observed criterion σ(I): 3.1 / Redundancy: 7.3 % / Biso Wilson estimate: 38.01 Å2 / Rmerge(I) obs: 0.116 / Net I/σ(I): 11.7 | |||||||||||||||
| Reflection shell | Highest resolution: 2.3 Å / Redundancy: 7.3 % / Rmerge(I) obs: 0.6 / Mean I/σ(I) obs: 11.7 / Num. unique all: 9999 / Rsym value: 0.605 / % possible all: 99.9 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.3→29.51 Å / Cor.coef. Fo:Fc: 0.946 / Cor.coef. Fo:Fc free: 0.901 / SU B: 20.582 / SU ML: 0.212 / Cross valid method: THROUGHOUT / σ(I): 3.1 / ESU R Free: 0.06 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.399 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→29.51 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Number: 663 / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Highest resolution: 2.3 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
