 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3txi: HEWL co-crystallization with carboplatin in DMSO media with parat... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3txi | ||||||
|---|---|---|---|---|---|---|---|
| Title | HEWL co-crystallization with carboplatin in DMSO media with paratone as the cryoprotectant | ||||||
 Components Components | Lysozyme C | ||||||
 Keywords Keywords | HYDROLASE / hen egg white lysozyme (HEWL) / bacterial cell wall lysis | ||||||
| Function / homology |  Function and homology information Function and homology informationLactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium ...Lactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium / defense response to Gram-positive bacterium / endoplasmic reticulum / : / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.6 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.6 Å | ||||||
 Authors Authors | Tanley, S.W.M. / Schreurs, A.M.M. / Helliwell, J.R. / Kroon-Batenburg, L.M.J. | ||||||
 Citation Citation | Journal: J.Appl.Crystallogr. / Year: 2013 Title: Experience with exchange and archiving of raw data: comparison of data from two diffractometers and four software packages on a series of lysozyme crystals. Authors: Tanley, S.W. / Schreurs, A.M. / Helliwell, J.R. / Kroon-Batenburg, L.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3txi.cif.gz | 69.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3txi.ent.gz | 50.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3txi.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tx/3txiftp://data.pdbj.org/pub/pdb/validation_reports/tx/3txi | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3txbC  3txdC  3txeC  3txfC  3txgC  3txhC  3txjC  3txkC  2w1yS  3txc 3txl C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |







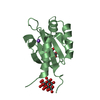



-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 14331.160 Da / Num. of mol.: 1 / Fragment: UNP residues 19-147 / Source method: isolated from a natural source / Source: (natural) |
|---|
-Non-polymers , 5 types, 139 molecules 

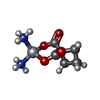


| #2: Chemical | ChemComp-CL /  Mass: 35.453 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Cl#3: Chemical | ChemComp-NA / |  Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#4: Chemical |  Mass: 371.248 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H12N2O4Pt / Comment: medication, chemotherapy*YM Mass: 371.248 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H12N2O4Pt / Comment: medication, chemotherapy*YM#5: Chemical | ChemComp-DMS /  Mass: 78.133 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 14 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 114 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.96 Å3/Da / Density % sol: 37.34 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: batch / pH: 4.7 Details: 49 mg HEWL + 3.7 mg carboplatin in 462.5 uL 0.04 M sodium acetate + 75 uL DMSO + 462.5 uL 10% sodium chloride, with paratone as cryoprotectant, pH 4.7, BATCH, temperature 295K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU MICROMAX-007 / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Jan 31, 2011 / Details: Osmic Confocal Max-Flux, blue configuration |
| Radiation | Monochromator: NONE / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→55.18 Å / Num. all: 15587 / Num. obs: 14573 / % possible obs: 93.5 % / Observed criterion σ(F): 4 / Observed criterion σ(I): 2 / Redundancy: 23.27 % / Biso Wilson estimate: 25.9 Å2 / Rmerge(I) obs: 0.054 / Rsym value: 0.054 / Net I/σ(I): 35.9 |
| Reflection shell | Resolution: 1.6→1.66 Å / Redundancy: 7.38 % / Rmerge(I) obs: 0.22 / Mean I/σ(I) obs: 6.2 / Num. unique all: 15587 / % possible all: 90.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2W1Y Resolution: 1.6→55.17 Å / Cor.coef. Fo:Fc: 0.954 / Cor.coef. Fo:Fc free: 0.931 / SU B: 3.839 / SU ML: 0.066 / Cross valid method: THROUGHOUT / σ(F): 4 / σ(I): 2 / ESU R: 0.107 / ESU R Free: 0.109 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : RESIDUAL ONLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.925 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→55.17 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.6→1.641 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -20.1003 Å / Origin y: -0.7753 Å / Origin z: -8.6806 Å
|
