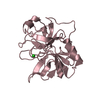
cytolytic granule / 加水分解酵素; プロテアーゼ; ペプチド結合加水分解酵素; セリンエンドペプチターゼ / Metabolism of Angiotensinogen to Angiotensins / protein maturation / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / killing of cells of another organism / serine-type endopeptidase activity / apoptotic process / proteolysis / extracellular space / membrane 類似検索 - 分子機能
Serine proteases, trypsin family, histidine active site / Serine proteases, trypsin family, serine active site / Serine proteases, trypsin family, histidine active site. / Peptidase S1A, chymotrypsin family / Serine proteases, trypsin family, serine active site. / Serine proteases, trypsin domain profile. / Trypsin-like serine protease / Serine proteases, trypsin domain / Trypsin / Trypsin-like serine proteases ...Serine proteases, trypsin family, histidine active site / Serine proteases, trypsin family, serine active site / Serine proteases, trypsin family, histidine active site. / Peptidase S1A, chymotrypsin family / Serine proteases, trypsin family, serine active site. / Serine proteases, trypsin domain profile. / Trypsin-like serine protease / Serine proteases, trypsin domain / Trypsin / Trypsin-like serine proteases / Thrombin, subunit H / Peptidase S1, PA clan, chymotrypsin-like fold / Peptidase S1, PA clan / Beta Barrel / Mainly Beta 類似検索 - ドメイン・相同性
1-acetyl-L-prolyl-L-threonyl-N-[(2R,3S)-4-chloro-3-hydroxy-1-(4-hydroxyphenyl)butan-2-yl]-L-serinamide / Granzyme H 類似検索 - 構成要素
THE UNBOUND FORM OF THE INHIBITOR (CHAIN B) IS ACE-PRO-THR-SER-TYR-CHLOROMETHYLKETONE. UPON ...THE UNBOUND FORM OF THE INHIBITOR (CHAIN B) IS ACE-PRO-THR-SER-TYR-CHLOROMETHYLKETONE. UPON REACTION WITH PROTEIN IT FORMS TWO COVALENT BONDS: 1) A COVALENT BOND TO SER 197 FORMING A HEMIACETAL AND 2) A COVALENT BOND TO NE2 OF HIS 59
解像度: 2.7→50 Å / Cor.coef. Fo:Fc: 0.912 / Cor.coef. Fo:Fc free: 0.885 / Occupancy max: 1 / Occupancy min: 0 / SU B: 29.31 / SU ML: 0.281 / 交差検証法: THROUGHOUT / ESU R: 0.862 / ESU R Free: 0.372 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD 詳細: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES
Rfactor
反射数
%反射
Selection details
Rfree
0.2885
406
4.7 %
RANDOM
Rwork
0.2294
8198
-
-
obs
0.232
8604
99.69 %
-
溶媒の処理
イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.4 Å / 溶媒モデル: MASK
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Homo sapiens (ヒト)
Homo sapiens (ヒト) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード










 PDBj
PDBj


 集合体
集合体

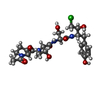
 タイプ: Peptide-like / クラス: 阻害剤 / 分子量: 527.011 Da / 分子数: 1 / 由来タイプ: 合成 / 詳細: This sequence is a synthetic construct.
タイプ: Peptide-like / クラス: 阻害剤 / 分子量: 527.011 Da / 分子数: 1 / 由来タイプ: 合成 / 詳細: This sequence is a synthetic construct.
 分子量: 96.063 Da / 分子数: 3 / 由来タイプ: 合成 / 式: SO4
分子量: 96.063 Da / 分子数: 3 / 由来タイプ: 合成 / 式: SO4 分子量: 18.015 Da / 分子数: 30 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 30 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: AR-NE3A / 波長: 1 Å
/ ビームライン: AR-NE3A / 波長: 1 Å 解析
解析