negative regulation of non-canonical inflammasome complex assembly / protein N-acetylglucosaminyltransferase complex / regulation of insulin receptor signaling pathway / protein O-GlcNAc transferase / protein O-acetylglucosaminyltransferase activity / positive regulation of transcription from RNA polymerase II promoter by glucose / acetylglucosaminyltransferase activity / regulation of Rac protein signal transduction / regulation of necroptotic process / protein O-linked glycosylation ...negative regulation of non-canonical inflammasome complex assembly / protein N-acetylglucosaminyltransferase complex / regulation of insulin receptor signaling pathway / protein O-GlcNAc transferase / protein O-acetylglucosaminyltransferase activity / positive regulation of transcription from RNA polymerase II promoter by glucose / acetylglucosaminyltransferase activity / regulation of Rac protein signal transduction / regulation of necroptotic process / protein O-linked glycosylation / negative regulation of stem cell population maintenance / Phosphorylation and nuclear translocation of BMAL1 (ARNTL) and CLOCK / regulation of gluconeogenesis / positive regulation of aggrephagy / regulation of chromosome separation / WNT mediated activation of DVL / Condensation of Prometaphase Chromosomes / regulation of glycolytic process / protein kinase CK2 complex / symbiont-mediated disruption of host cell PML body / RIPK1-mediated regulated necrosis / Phosphorylation and nuclear translocation of the CRY:PER:kinase complex / Regulation of CDH1 posttranslational processing and trafficking to plasma membrane / Receptor Mediated Mitophagy / Sin3-type complex / regulation of synapse assembly / Formation of WDR5-containing histone-modifying complexes / positive regulation of stem cell population maintenance / Synthesis of PC / hemopoiesis / regulation of neurotransmitter receptor localization to postsynaptic specialization membrane / positive regulation of proteolysis / negative regulation of signal transduction by p53 class mediator / phosphatidylinositol-3,4,5-trisphosphate binding / RUNX1 interacts with co-factors whose precise effect on RUNX1 targets is not known / negative regulation of apoptotic signaling pathway / Maturation of hRSV A proteins / histone acetyltransferase complex / negative regulation of double-strand break repair via homologous recombination / positive regulation of Wnt signaling pathway / mitophagy / cell projection / response to nutrient / positive regulation of TORC1 signaling / negative regulation of protein ubiquitination / negative regulation of proteasomal ubiquitin-dependent protein catabolic process / negative regulation of cell migration / Signal transduction by L1 / positive regulation of translation / circadian regulation of gene expression / cellular response to glucose stimulus / negative regulation of transforming growth factor beta receptor signaling pathway / protein processing / response to insulin / Hsp90 protein binding / chromatin DNA binding / PML body / Wnt signaling pathway / Regulation of necroptotic cell death / mitochondrial membrane / Regulation of PTEN stability and activity / kinase activity / positive regulation of protein catabolic process / UCH proteinases / KEAP1-NFE2L2 pathway / rhythmic process / double-strand break repair / Cooperation of PDCL (PhLP1) and TRiC/CCT in G-protein beta folding / positive regulation of cold-induced thermogenesis / HATs acetylate histones / positive regulation of cell growth / chromatin organization / protein folding / Regulation of TP53 Activity through Phosphorylation / non-specific serine/threonine protein kinase / regulation of cell cycle / negative regulation of translation / protein stabilization / protein serine kinase activity / protein serine/threonine kinase activity / positive regulation of cell population proliferation / apoptotic process / regulation of transcription by RNA polymerase II / DNA damage response / positive regulation of DNA-templated transcription / glutamatergic synapse / negative regulation of transcription by RNA polymerase II / signal transduction / positive regulation of transcription by RNA polymerase II / protein-containing complex / nucleoplasm / ATP binding / identical protein binding / nucleus / plasma membrane / cytosol Similarity search - Function
Signal recognition particle alu RNA binding heterodimer, srp9/1 - #150 / UDP-N-acetylglucosamine--peptide N-acetylglucosaminyltransferase 110kDa subunit / Rossmann fold - #11380 / O-GlcNAc transferase, C-terminal / Glycosyl transferase family 41 / Signal recognition particle alu RNA binding heterodimer, srp9/1 / TPR repeat / Tetratricopeptide repeat / Tetratricopeptide repeat / Casein Kinase 2, subunit alpha ...Signal recognition particle alu RNA binding heterodimer, srp9/1 - #150 / UDP-N-acetylglucosamine--peptide N-acetylglucosaminyltransferase 110kDa subunit / Rossmann fold - #11380 / O-GlcNAc transferase, C-terminal / Glycosyl transferase family 41 / Signal recognition particle alu RNA binding heterodimer, srp9/1 / TPR repeat / Tetratricopeptide repeat / Tetratricopeptide repeat / Casein Kinase 2, subunit alpha / Tetratricopeptide repeat domain / Tetratricopeptide repeat / Glycogen Phosphorylase B; / TPR repeat region circular profile. / TPR repeat profile. / Tetratricopeptide repeats / Tetratricopeptide repeat / Serine Threonine Protein Phosphatase 5, Tetratricopeptide repeat / Alpha Horseshoe / Tetratricopeptide-like helical domain superfamily / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / Rossmann fold / 2-Layer Sandwich / 3-Layer(aba) Sandwich / Mainly Alpha / Alpha Beta Similarity search - Domain/homology
FORMYL GROUP / URIDINE-5'-DIPHOSPHATE / UDP-N-acetylglucosamine--peptide N-acetylglucosaminyltransferase 110 kDa subunit / Casein kinase II subunit alpha Similarity search - Component
Mass: 18.015 Da / Num. of mol.: 1018 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has protein modification
Y
Nonpolymer details
THE INHIBITOR, 4-METHOXYPHENYL 6-ACETYL-2-BENZOXAZOLINONE- CARBOXYLATE, CROSSLINKS THE ACTIVE SITE ...THE INHIBITOR, 4-METHOXYPHENYL 6-ACETYL-2-BENZOXAZOLINONE- CARBOXYLATE, CROSSLINKS THE ACTIVE SITE OF OGT. EXPERIMENTAL EVIDENCE SUGGESTS THAT LYSINE 842 REACTS WITH ONE OF THE TWO CARBAMATES OF THE INHIBITOR, RELEASING 6-ACETYL-2-BENZOXAZOLINONE. THEN, THE NEARBY CYSTEINE 917 REACTS AGAIN WITH THE LYSINE ADDUCT TO RELEASE 4-METHOXYPHENOL AND GENERATES A COVALENT THIOCARBAMATE LINKAGE BETWEEN LYSINE 842 AND CYSTEINE 917.
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 3.05 Å3/Da / Density % sol: 59.71 %
Crystal grow
Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 1.6M Lithium Sulfate, 0.1M Bis Tris Propane pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 298.0K
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj









 Assembly
Assembly





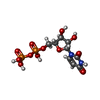



 Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM
Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM Mass: 30.026 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH2O
Mass: 30.026 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CH2O Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Sample preparation
Sample preparation / Beamline: X25 / Wavelength: 1 Å
/ Beamline: X25 / Wavelength: 1 Å Processing
Processing