 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3s3e: Crystal structure of the catalytic domain of PTP10D from Drosophi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3s3e | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the catalytic domain of PTP10D from Drosophila melanogaster | ||||||
 Components Components | Tyrosine-protein phosphatase 10D | ||||||
 Keywords Keywords | HYDROLASE / Differentiation / Neurogenesis / Signal transduction / Developmental Protein / Protein Phosphatase / Protein Tyrosine Phosphatase | ||||||
| Function / homology |  Function and homology information Function and homology informationmembrane-membrane adaptor activity / branching involved in open tracheal system development / cell-cell signaling via exosome / open tracheal system development / negative regulation of fibroblast growth factor receptor signaling pathway / transmembrane receptor protein tyrosine phosphatase activity / Neutrophil degranulation / motor neuron axon guidance / negative regulation of vascular endothelial growth factor receptor signaling pathway / negative regulation of epidermal growth factor receptor signaling pathway ...membrane-membrane adaptor activity / branching involved in open tracheal system development / cell-cell signaling via exosome / open tracheal system development / negative regulation of fibroblast growth factor receptor signaling pathway / transmembrane receptor protein tyrosine phosphatase activity / Neutrophil degranulation / motor neuron axon guidance / negative regulation of vascular endothelial growth factor receptor signaling pathway / negative regulation of epidermal growth factor receptor signaling pathway / long-term memory / protein-tyrosine-phosphatase / protein tyrosine phosphatase activity / axon guidance / central nervous system development / apical part of cell / axon / signal transduction / membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Madan, L.L. / Gopal, B. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2011 Title: Conformational basis for substrate recruitment in protein tyrosine phosphatase 10D Authors: Madan, L.L. / Gopal, B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3s3e.cif.gz | 243.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3s3e.ent.gz | 197.2 KB | Display | PDB format |
| PDBx/mmJSON format | 3s3e.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/s3/3s3eftp://data.pdbj.org/pub/pdb/validation_reports/s3/3s3e | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3s3fC  3s3hC  3s3kC  2ahsS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj











- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||
| 2 | 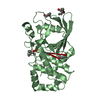
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Ens-ID: 1 / Beg auth comp-ID: MET / Beg label comp-ID: MET / End auth comp-ID: LYS / End label comp-ID: LYS / Refine code: 5 / Auth seq-ID: 21 - 305 / Label seq-ID: 21 - 305
|
-Components
| #1: Protein | Mass: 35945.633 Da / Num. of mol.: 2 / Fragment: UNP residues 1250-1533 Source method: isolated from a genetically manipulated source Details: Central nervous system / Source: (gene. exp.)  #2: Chemical |   Mass: 60.095 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O Mass: 60.095 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O#3: Chemical | ChemComp-1BO / 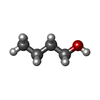  Mass: 74.122 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C4H10O Mass: 74.122 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C4H10O#4: Chemical | ChemComp-BU1 / | 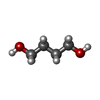  Mass: 90.121 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O2 Mass: 90.121 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 160 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 160 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.62 Å3/Da / Density % sol: 66.04 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: 15% PEG 4000, 120mM Citrate, 10% iso-propanol, 10% n-Butanol, 10% 1,4-Butanediol , pH 6.5, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 0.95 Å / Beamline: BM14 / Wavelength: 0.95 Å |
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Apr 21, 2010 / Details: bent collimating mirror and toroid |
| Radiation | Monochromator: Si 111 CHANNEL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.95 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→44 Å / Num. obs: 41551 / % possible obs: 100 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -5 / Redundancy: 11.2 % / Biso Wilson estimate: 38.12 Å2 / Rmerge(I) obs: 0.119 / Net I/σ(I): 15.9 |
| Reflection shell | Resolution: 2.4→2.53 Å / Redundancy: 11.3 % / Rmerge(I) obs: 0.484 / Mean I/σ(I) obs: 5.2 / Num. unique all: 67497 / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2AHS Resolution: 2.4→44 Å / Cor.coef. Fo:Fc: 0.928 / Cor.coef. Fo:Fc free: 0.91 / SU B: 12.185 / SU ML: 0.13 / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / ESU R Free: 0.196 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.751 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→44 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Dom-ID: 1 / Auth asym-ID: A / Ens-ID: 1 / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.462 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
