 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
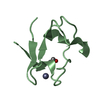
Yorodumi- PDB-3rzt: Neutron structure of perdeuterated rubredoxin using rapid (14 hou... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3rzt | ||||||
|---|---|---|---|---|---|---|---|
| Title | Neutron structure of perdeuterated rubredoxin using rapid (14 hours) data | ||||||
 Components Components | Rubredoxin | ||||||
 Keywords Keywords | ELECTRON TRANSPORT / Iron / metal-binding / transport | ||||||
| Function / homology |  Function and homology information Function and homology informationalkane catabolic process / electron transfer activity / iron ion binding Similarity search - Function | ||||||
| Biological species |   Pyrococcus furiosus (archaea) Pyrococcus furiosus (archaea) | ||||||
| Method | NEUTRON DIFFRACTION / NUCLEAR REACTOR /  FOURIER SYNTHESIS / Resolution: 1.7504 Å FOURIER SYNTHESIS / Resolution: 1.7504 Å | ||||||
 Authors Authors | Munshi, P. / Chung, C.-L. / Weiss, K.L. / Blakeley, M.P. / Myles, D.A.A. / Meilleur, F. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2012 Title: Rapid visualization of hydrogen positions in protein neutron crystallographic structures. Authors: Munshi, P. / Chung, S.L. / Blakeley, M.P. / Weiss, K.L. / Myles, D.A. / Meilleur, F. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3rzt.cif.gz | 31.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3rzt.ent.gz | 20.8 KB | Display | PDB format |
| PDBx/mmJSON format | 3rzt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rz/3rztftp://data.pdbj.org/pub/pdb/validation_reports/rz/3rzt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3rygC  3rz6C  3ss2C  3kywS  3rzr C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 6031.728 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pyrococcus furiosus (archaea) / Strain: ATCC 43587 / DSM 3638 / JCM 8422 / Vc1 / Gene: PF1282, rub / Plasmid: PET24D / Production host:  |
|---|---|
| #2: Chemical | ChemComp-FE /   Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe |
| #3: Chemical | ChemComp-DOD /   Mass: 18.015 Da / Num. of mol.: 35 / Source method: isolated from a natural source / Formula: D2O Mass: 18.015 Da / Num. of mol.: 35 / Source method: isolated from a natural source / Formula: D2O |
-Experimental details
-Experiment
| Experiment | Method: NEUTRON DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: 20 mg/ml rubredoxin in 1.9 M sodium/potassium phosphate sitting drop equilibrated against 3.8 m sodium/potassium phosphate reservoir, VAPOR DIFFUSION, SITTING DROP, temperature 293K |
|---|
-Data collection
| Diffraction | Mean temperature: 293 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: NUCLEAR REACTOR / Site: ILL  / Beamline: LADI III / Wavelength: 3.2 - 4.2 / Beamline: LADI III / Wavelength: 3.2 - 4.2 | |||||||||
| Detector | Type: MAATEL QLD: LADI-III / Detector: IMAGE PLATE / Date: Jun 8, 2007 | |||||||||
| Radiation | Protocol: LAUE / Monochromatic (M) / Laue (L): L / Scattering type: neutron | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 1.75→27.21 Å / Num. all: 4188 / Num. obs: 4187 / % possible obs: 77.5 % / Observed criterion σ(I): 2 / Redundancy: 4.6 % / Rmerge(I) obs: 0.153 / Net I/σ(I): 8.4 | |||||||||
| Reflection shell | Resolution: 1.75→1.84 Å / Redundancy: 2.6 % / Rmerge(I) obs: 0.189 / Mean I/σ(I) obs: 3.6 / % possible all: 56.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: PDB ENTRY 3KYW Resolution: 1.7504→27.208 Å / SU ML: 0.44 / σ(F): 1.53 / Phase error: 21.95 / Stereochemistry target values: ML
| ||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 1.2 Å / VDW probe radii: 1 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 49.505 Å2 / ksol: 0.6 e/Å3 | ||||||||||||||||||||||||
| Displacement parameters |
| ||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7504→27.208 Å
| ||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.7504→27.2108 Å
|
