 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3req: METHYLMALONYL-COA MUTASE, SUBSTRATE-FREE STATE (POOR QUALITY STRU... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3req | ||||||
|---|---|---|---|---|---|---|---|
| Title | METHYLMALONYL-COA MUTASE, SUBSTRATE-FREE STATE (POOR QUALITY STRUCTURE) | ||||||
 Components Components | (METHYLMALONYL-COA MUTASE) x 2 | ||||||
 Keywords Keywords | COMPLEX (ISOMERASE/DEOXYADENOSINE) / COMPLEX (ISOMERASE-DEOXYADENOSINE) / ISOMERASE / MUTASE / INTRAMOLECULAR TRANSFERASE / COMPLEX (ISOMERASE-DEOXYADENOSINE) complex | ||||||
| Function / homology |  Function and homology information Function and homology information: / : / methylmalonyl-CoA mutase / methylmalonyl-CoA mutase activity / cobalamin binding / metal ion binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  Propionibacterium freudenreichii subsp. shermanii (bacteria) Propionibacterium freudenreichii subsp. shermanii (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.7 Å | ||||||
 Authors Authors | Evans, P.R. / Mancia, F. | ||||||
 Citation Citation | Journal: Structure / Year: 1998 Title: Conformational changes on substrate binding to methylmalonyl CoA mutase and new insights into the free radical mechanism. Authors: Mancia, F. / Evans, P.R. #1: Journal: Structure / Year: 1996Title: How Coenzyme B12 Radicals are Generated: The Crystal Structure of Methylmalonyl-Coenzyme a Mutase at 2 A Resolution Authors: Mancia, F. / Keep, N.H. / Nakagawa, A. / Leadlay, P.F. / Mcsweeney, S. / Rasmussen, B. / Bosecke, P. / Diat, O. / Evans, P.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3req.cif.gz | 277.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3req.ent.gz | 214.4 KB | Display | PDB format |
| PDBx/mmJSON format | 3req.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/re/3reqftp://data.pdbj.org/pub/pdb/validation_reports/re/3req | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2reqSC  4reqC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | THE CRYSTAL ASYMMETRIC UNIT CONSISTS OF TWO HETERODIMERIC MOLECULES, EACH WITH AN ACTIVE ALPHA CHAIN (CHAINS A AND C), AND AN INACTIVE BETA CHAIN (CHAINS B AND D). EACH ALPHA CHAIN CONTAINS A COBALAMIN, MODELED AS A FIVE-COORDINATE CO (II) SPECIES, AND A COENZYME A MOLECULE APPROXIMATELY IN THE SUBSTRATE BINDING SITE, BUT WITH THE PANTOTHEINE SIDE CHAIN DISORDERED. |
-Components
| #1: Protein | Mass: 80137.852 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Propionibacterium freudenreichii subsp. shermanii (bacteria)Species: Propionibacterium freudenreichii / Strain: NCIB 9885 Description: THE 2 GENES MUTA (BETA CHAIN) AND MUTB (ALPHA CHAIN) ARE COEXPRESSED FROM THE SAME PLASMID Gene: MUTA, MUTB / Plasmid: PMEX2 / Cellular location (production host): CYTOPLASM / Gene (production host): MUTA, MUTB / Production host: |
|---|---|
| #2: Protein | Mass: 69430.188 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Propionibacterium freudenreichii subsp. shermanii (bacteria)Species: Propionibacterium freudenreichii / Strain: NCIB 9885 Description: THE 2 GENES MUTA (BETA CHAIN) AND MUTB (ALPHA CHAIN) ARE COEXPRESSED FROM THE SAME PLASMID Gene: MUTA, MUTB / Plasmid: PMEX2 / Cellular location (production host): CYTOPLASM / Gene (production host): MUTA, MUTB / Production host: K38 PGP1-2 / References: UniProt: P11652, methylmalonyl-CoA mutase |
| #3: Chemical | ChemComp-B12 / 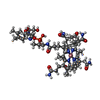  Mass: 1330.356 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C62H89CoN13O14P Mass: 1330.356 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C62H89CoN13O14P |
| #4: Chemical | ChemComp-ADN / 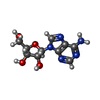  Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 Mass: 267.241 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H13N5O4 |
| Compound details | THIS STRUCTURE IS IN THE OPEN SUBSTRATE-FREE CONFORMATION. THE PROTEIN CONFORMATION IS VERY SIMILAR ...THIS STRUCTURE IS IN THE OPEN SUBSTRATE-FREE CONFORMATI |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 53 % Description: THE DATA WERE COLLECTED IN 3 PASSES OF DIFFERENT EXPOSURE TIMES BECAUSE OF THE VERY HIGH BFACTOR. THE 3 PASSES MERGED TOGETHER VERY POORLY. |
|---|---|
| Crystal grow | Method: vapor diffusion / pH: 7.5 Details: PROTEIN SOLUTION: 20 MG/ML PROTEIN, 1MM ADENOSYLCOBALAMIN, 1MM DTT, TRIS PH 7.5. RESERVOIR: 14% PEG 4000 (W/V), 20% GLYCEROL (V/V), 100MM TRIS-HCL PH 7.5. EQUAL VOLUMES OF PROTEIN SOLUTION ...Details: PROTEIN SOLUTION: 20 MG/ML PROTEIN, 1MM ADENOSYLCOBALAMIN, 1MM DTT, TRIS PH 7.5. RESERVOIR: 14% PEG 4000 (W/V), 20% GLYCEROL (V/V), 100MM TRIS-HCL PH 7.5. EQUAL VOLUMES OF PROTEIN SOLUTION AND RESERVOIR MIXED, AND EQUILIBRATED BY VAPOR DIFFUSION., vapor diffusion |
| Crystal grow | *PLUS Method: unknown |
| Components of the solutions | *PLUS Conc.: 14 % / Common name: PEG |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 1 / Beamline: BM14 / Wavelength: 1 |
| Detector | Type: MAR scanner 300 mm plate / Detector: IMAGE PLATE / Date: Jun 1, 1996 / Details: 2 MIRRORS, 2 SI(111) CRYSTAL MONOCHROMATOR |
| Radiation | Monochromator: SI(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→29 Å / Num. obs: 44606 / % possible obs: 99.2 % / Observed criterion σ(I): 6 / Redundancy: 9 % / Biso Wilson estimate: 78 Å2 / Rmerge(I) obs: 0.072 / Net I/σ(I): 7.9 |
| Reflection shell | Resolution: 2.7→2.85 Å / Redundancy: 6.8 % / Rmerge(I) obs: 0.381 / Mean I/σ(I) obs: 2 / % possible all: 98.7 |
| Reflection shell | *PLUS % possible obs: 99.2 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2REQ Resolution: 2.7→20 Å / Cross valid method: THROUGHOUT / σ(F): 0 / Details: DERIVED FROM ENGH AND HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 68 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.313 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 2.7 Å / Lowest resolution: 2.85 Å |
