 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3r0w: Crystal Structures of Multidrug-resistant HIV-1 Protease in Compl... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3r0w | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structures of Multidrug-resistant HIV-1 Protease in Complex with Mechanism-Based Aspartyl Protease Inhibitors. | ||||||
 Components Components | Multidrug-resistant clinical isolate 769 HIV-1 Protease | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationviral genome integration into host DNA / establishment of integrated proviral latency / RNA stem-loop binding / host multivesicular body / RNA-directed DNA polymerase activity / endonuclease activity / aspartic-type endopeptidase activity / virion membrane / proteolysis / DNA binding / zinc ion binding Similarity search - Function | ||||||
| Biological species |   Human immunodeficiency virus 1 Human immunodeficiency virus 1 | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Yedidi, R.S. / Gupta, D. / Liu, Z. / Brunzelle, J. / Kovari, I.A. / Woster, P.M. / Kovari, L.C. | ||||||
 Citation Citation | Journal: Biochem.Biophys.Res.Commun. / Year: 2012 Title: Crystal structures of multidrug-resistant HIV-1 protease in complex with two potent anti-malarial compounds. Authors: Yedidi, R.S. / Liu, Z. / Wang, Y. / Brunzelle, J.S. / Kovari, I.A. / Woster, P.M. / Kovari, L.C. / Gupta, D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3r0w.cif.gz | 57.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3r0w.ent.gz | 40.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3r0w.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/r0/3r0wftp://data.pdbj.org/pub/pdb/validation_reports/r0/3r0w | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3r0yC  1tw7S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 10769.635 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Human immunodeficiency virus 1 / Production host:  #2: Chemical | ChemComp-RSY / | 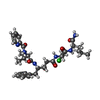  Type: peptide-like, Peptide-like / Class: Enzyme inhibitor / Mass: 631.163 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H43ClN6O6 Type: peptide-like, Peptide-like / Class: Enzyme inhibitor / Mass: 631.163 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H43ClN6O6References: N-[(2R)-1-{[(2S,3S)-5-{[(2R)-1-{[(2S)-1-AMINO-4-METHYL-1-OXOPENTAN-2-YL]AMINO}-3-CHLORO-1-OXOPROPAN-2-YL]AMINO}-3-HYDROXY-5-OXO-1-PHENYLPENTAN-2-YL]AMINO}-3-METHYL-1-OXOBUTAN-2-YL]PYRIDINE-2-CARBOXAMIDE #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 208 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.48 Å3/Da / Density % sol: 50.38 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.2 Details: 0.3 M NaCl, 0.1 M HEPES, pH 7.2, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 21-ID-F / Wavelength: 1 Å / Beamline: 21-ID-F / Wavelength: 1 Å |
| Detector | Type: MAR CCD 165 mm / Detector: CCD / Date: Sep 4, 2008 |
| Radiation | Monochromator: mirrors and beryllium lenses / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→50 Å / Num. obs: 22795 / Rmerge(I) obs: 0.04 |
| Reflection shell | Resolution: 1.7→1.76 Å / % possible all: 100 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1TW7 Resolution: 1.7→18.98 Å / Cor.coef. Fo:Fc: 0.955 / Cor.coef. Fo:Fc free: 0.934 / SU B: 2.082 / SU ML: 0.072 / Cross valid method: THROUGHOUT / ESU R Free: 0.122 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: 1) DUE TO MULTIPLE BINDING ORIENTATIONS OF THE LIGAND, THE DIFFERENCE ELECTRON DENSITY (CALCULATED USING XTALVIEW/XFIT PROGRAM) WAS CONTOURED BETWEEN 1.8 AND 2.0 SIGMA WHILE FITTING THE ...Details: 1) DUE TO MULTIPLE BINDING ORIENTATIONS OF THE LIGAND, THE DIFFERENCE ELECTRON DENSITY (CALCULATED USING XTALVIEW/XFIT PROGRAM) WAS CONTOURED BETWEEN 1.8 AND 2.0 SIGMA WHILE FITTING THE LIGAND. EACH OF THE CONFORMATIONS WAS MANUALLY FIT INTO THE DENSITY AND REFINED USING REFMAC5. REFMAC LIBRARY FOR THE LIGAND WAS GENERATED USING MONOMER LIBRARY SKETCHER OPTION IN CCP4 AS WELL AS OBTAINED FROM PRODRG SERVER. REFINED COORDINATES FOR THE LIGAND CONFORMATION WITH HIGHEST OCCUPANCY AND REASONABLE THERMAL FACTOR VALUES ARE INCLUDED IN THE FINAL COORDINATE FILE. 2) HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.058 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→18.98 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.7→1.746 Å / Total num. of bins used: 20
|
