 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3pec: Siderocalin Recognitin of Carboxymycobactins: Interference by the... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3pec | ||||||
|---|---|---|---|---|---|---|---|
| Title | Siderocalin Recognitin of Carboxymycobactins: Interference by the immune system in intracellular iron acquisition by Mycobacteria tuberculosis | ||||||
 Components Components | Neutrophil gelatinase-associated lipocalin | ||||||
 Keywords Keywords | ANTIMICROBIAL PROTEIN / 8-stranded anti-parallel beta barrel / Siderocalin / Siderophore binding | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of iron ion import across plasma membrane / positive regulation of hippocampal neuron apoptotic process / positive regulation of endothelial tube morphogenesis / negative regulation of hippocampal neuron apoptotic process / positive regulation of cell projection organization / Metal sequestration by antimicrobial proteins / response to mycotoxin / response to kainic acid / siderophore transport / cellular response to increased oxygen levels ...positive regulation of iron ion import across plasma membrane / positive regulation of hippocampal neuron apoptotic process / positive regulation of endothelial tube morphogenesis / negative regulation of hippocampal neuron apoptotic process / positive regulation of cell projection organization / Metal sequestration by antimicrobial proteins / response to mycotoxin / response to kainic acid / siderophore transport / cellular response to increased oxygen levels / response to blue light / response to fructose / cellular response to X-ray / short-term memory / cellular response to interleukin-6 / enterobactin binding / iron ion sequestering activity / response to herbicide / response to iron(II) ion / positive regulation of reactive oxygen species biosynthetic process / cellular response to interleukin-1 / long-term memory / extrinsic apoptotic signaling pathway in absence of ligand / positive regulation of endothelial cell migration / cellular response to nutrient levels / acute-phase response / cellular response to nerve growth factor stimulus / Iron uptake and transport / cellular response to tumor necrosis factor / specific granule lumen / cellular response to amyloid-beta / cellular response to hydrogen peroxide / response to virus / positive regulation of cold-induced thermogenesis / cellular response to lipopolysaccharide / protease binding / Interleukin-4 and Interleukin-13 signaling / cellular response to hypoxia / defense response to bacterium / response to xenobiotic stimulus / iron ion binding / innate immune response / positive regulation of gene expression / Neutrophil degranulation / : / extracellular exosome / extracellular region / identical protein binding Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Used previously-determined structure / Resolution: 2.19 Å X-RAY DIFFRACTION / SYNCHROTRON / Used previously-determined structure / Resolution: 2.19 Å | ||||||
 Authors Authors | Clifton, M.C. | ||||||
 Citation Citation | Journal: To be Published Title: Siderocalin Recognitin of Carboxymycobactins: Interference by the immune system in intracellular iron acquisition by Mycobacteria tuberculosis Authors: Hoette, T.M. / Clifton, M.C. / Zawadzka, A.M. / Holmes, M.A. / Strong, R.K. / Raymond, K.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3pec.cif.gz | 224.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3pec.ent.gz | 181.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3pec.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pe/3pecftp://data.pdbj.org/pub/pdb/validation_reports/pe/3pec | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3pedC  1l6mS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 3 molecules ABC
| #1: Protein | Mass: 20556.438 Da / Num. of mol.: 3 / Fragment: UNP residues 21 to 198 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: LCN2, HNL, NGAL / Production host:  |
|---|
-Non-polymers , 6 types, 244 molecules 



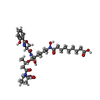

| #2: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: C3H8O3#3: Chemical |  Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-FE / |  Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe Mass: 55.845 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Fe#5: Chemical | ChemComp-NA /  Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na#6: Chemical | ChemComp-ZYG / |  Mass: 735.822 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C35H53N5O12 Mass: 735.822 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C35H53N5O12#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 228 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has protein modification | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.15 Å3/Da / Density % sol: 61 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 4.5 Details: 1.3M Ammonium sulfate, 0.2M Lithium sulfate, 50mM sodium chloride, 0.1M sodium acetate, pH 4.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.1 / Wavelength: 0.97 Å / Beamline: 5.0.1 / Wavelength: 0.97 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Apr 1, 2009 |
| Radiation | Monochromator: Single crystal, cylindrically bent, Si(220) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→50 Å / Num. all: 77333 / Num. obs: 41091 / % possible obs: 100 % |
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 7 % / Rmerge(I) obs: 0.35 / Mean I/σ(I) obs: 5.6 / Num. unique all: 4024 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: Used previously-determined structure Starting model: PDB entry 1L6M Resolution: 2.19→40.66 Å / Cor.coef. Fo:Fc: 0.923 / Cor.coef. Fo:Fc free: 0.91 / SU B: 11.636 / SU ML: 0.139 / Cross valid method: THROUGHOUT / ESU R Free: 0.212 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 44.53 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.19→40.66 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.186→2.243 Å / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
