RNA exonuclease activity / DNA ligase (ATP) / DNA ligase (ATP) activity / DNA replication, synthesis of primer / DNA biosynthetic process / nucleotidyltransferase activity / double-strand break repair via nonhomologous end joining / DNA recombination / DNA-directed DNA polymerase activity / DNA binding ...RNA exonuclease activity / DNA ligase (ATP) / DNA ligase (ATP) activity / DNA replication, synthesis of primer / DNA biosynthetic process / nucleotidyltransferase activity / double-strand break repair via nonhomologous end joining / DNA recombination / DNA-directed DNA polymerase activity / DNA binding / ATP binding / metal ion binding Similarity search - Function
DNA ligase D / LigD polymerase domain, PaeLigD-type / DNA ligase D, ligase domain / DNA ligase D, polymerase domain / : / LigD, primase-polymerase domain / DNA ligase D, 3'-phosphoesterase domain / DNA Ligase D 3'-phosphoesterase domain / DNA ligase, ATP-dependent, C-terminal / ATP dependent DNA ligase C terminal region ...DNA ligase D / LigD polymerase domain, PaeLigD-type / DNA ligase D, ligase domain / DNA ligase D, polymerase domain / : / LigD, primase-polymerase domain / DNA ligase D, 3'-phosphoesterase domain / DNA Ligase D 3'-phosphoesterase domain / DNA ligase, ATP-dependent, C-terminal / ATP dependent DNA ligase C terminal region / ATP-dependent DNA ligase family profile. / DNA ligase, ATP-dependent, central / ATP dependent DNA ligase domain / Nucleic acid-binding, OB-fold Similarity search - Domain/homology
: / DI(HYDROXYETHYL)ETHER / YTTRIUM (III) ION / Multifunctional non-homologous end joining protein LigD Similarity search - Component
Biological species
Pseudomonas aeruginosa (bacteria)
Method
X-RAY DIFFRACTION / SYNCHROTRON / MIR / Resolution: 1.92 Å
Mass: 18.015 Da / Num. of mol.: 358 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has protein modification
N
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.24 Å3/Da / Density % sol: 45.09 %
Crystal grow
Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: Crystallization was carried out in sitting-drop vapor-diffusion setups with 1:1 mixtures of protein solution containing 1.3 mM PaePEC2 and 2mM MnCl2 and reservoir solution containing PEG ...Details: Crystallization was carried out in sitting-drop vapor-diffusion setups with 1:1 mixtures of protein solution containing 1.3 mM PaePEC2 and 2mM MnCl2 and reservoir solution containing PEG 5000 monomethylether (MME) (20 - 30%), 100 mM 2-(N-morpholino) ethanesulfonic acid (MES) pH 6.8 - 7.0, 200 mM ammonium sulfate, and 10 mM yttrium (III) chloride at 22 C. , VAPOR DIFFUSION, SITTING DROP, temperature 298K
-
Data collection
Diffraction
ID
Mean temperature (K)
Crystal-ID
1
130
1
2
1
3
1
Diffraction source
Source
Site
Beamline
Type
ID
Wavelength (Å)
SYNCHROTRON
NSLS
X12C
1
1.19, 0.99, 0.73
SYNCHROTRON
NSLS
X25
2
1.19, 0.99, 0.73
ROTATING ANODE
RIGAKU RU300
3
1.54
Detector
Type
ID
Detector
Date
Details (eV)
ADSC QUANTUM 315
1
CCD
Feb 20, 2010
SeeNSLSX25BeamlineDescription
ADSC QUANTUM 210
2
CCD
Feb 20, 2010
SeeNSLSX12CBeamlineDescription
RIGAKU RAXIS IV++
3
IMAGE PLATE
Feb 10, 2010
Osmicconfocalmirrors
Radiation
Monochromator: See Beamline Documentation / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
ID
Wavelength (Å)
Relative weight
1
1.19
1
2
0.99
1
3
0.73
1
4
1.54
1
Reflection
Resolution: 1.92→50 Å / Num. all: 25728 / Num. obs: 24596 / % possible obs: 95.6 % / Observed criterion σ(I): -3 / Redundancy: 7.4 % / Biso Wilson estimate: 26 Å2 / Rsym value: 0.087 / Net I/σ(I): 25.9
Reflection shell
Resolution: 1.92→1.95 Å / Redundancy: 2.1 % / Mean I/σ(I) obs: 4.8 / Num. unique all: 1272 / % possible all: 93
-
Processing
Software
Name
Version
Classification
HKL-2000
datacollection
SOLVE
phasing
PHENIX
(phenix.refine: 1.6_289)
refinement
HKL-2000
datareduction
SCALEPACK
datascaling
Refinement
Method to determine structure: MIR / Resolution: 1.92→37.395 Å / SU ML: 0.2 / σ(F): 0.06 / Stereochemistry target values: ML
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.1988
2387
9.95 %
10% selected at random in CCP4/Unique
Rwork
0.1475
-
-
-
obs
0.1527
23992
92.95 %
-
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 53.778 Å2 / ksol: 0.344 e/Å3
Displacement parameters
Baniso -1
Baniso -2
Baniso -3
1-
5.3212 Å2
-1.2709 Å2
-1.1842 Å2
2-
-
-1.6351 Å2
0.4754 Å2
3-
-
-
-3.7302 Å2
Refinement step
Cycle: LAST / Resolution: 1.92→37.395 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
2439
0
34
358
2831
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.01
2617
X-RAY DIFFRACTION
f_angle_d
1.249
3556
X-RAY DIFFRACTION
f_dihedral_angle_d
14.969
993
X-RAY DIFFRACTION
f_chiral_restr
0.1
370
X-RAY DIFFRACTION
f_plane_restr
0.005
463
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
1.92-1.9886
0.2438
208
0.1666
1912
X-RAY DIFFRACTION
82
1.9886-2.0682
0.2206
241
0.1376
2149
X-RAY DIFFRACTION
92
2.0682-2.1624
0.2045
239
0.1386
2170
X-RAY DIFFRACTION
94
2.1624-2.2763
0.2545
200
0.1725
1597
X-RAY DIFFRACTION
70
2.2763-2.4189
0.2095
244
0.162
2195
X-RAY DIFFRACTION
94
2.4189-2.6057
0.2057
263
0.1515
2272
X-RAY DIFFRACTION
99
2.6057-2.8678
0.2094
223
0.1598
2353
X-RAY DIFFRACTION
100
2.8678-3.2826
0.1962
247
0.1507
2340
X-RAY DIFFRACTION
100
3.2826-4.1348
0.1807
254
0.1239
2325
X-RAY DIFFRACTION
99
4.1348-37.4023
0.1645
268
0.1364
2292
X-RAY DIFFRACTION
100
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Pseudomonas aeruginosa (bacteria)
Pseudomonas aeruginosa (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
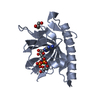









 PDBj
PDBj


 Assembly
Assembly








 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3
Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn
Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Mass: 88.906 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Y
Mass: 88.906 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Y Sample preparation
Sample preparation
 Processing
Processing