 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3mhj: Human tankyrase 2 - catalytic PARP domain in complex with 1-methy... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3mhj | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human tankyrase 2 - catalytic PARP domain in complex with 1-methyl-3-(trifluoromethyl)-5h-benzo[c][1,8]naphtyridine-6-one | ||||||
 Components Components | Tankyrase-2 | ||||||
 Keywords Keywords | TRANSFERASE / CATALYTIC FRAGMENT / PARP / STRUCTURAL GENOMICS / STRUCTURAL GENOMICS CONSORTIUM / SGC / ADP-RIBOSYLATION / ANK REPEAT / CHROMOSOMAL PROTEIN / GLYCOSYLTRANSFERASE / GOLGI APPARATUS / MEMBRANE / MRNA TRANSPORT / NAD / NUCLEUS / PHOSPHORYLATION / PROTEIN TRANSPORT / TELOMERE / TRANSLOCATION / TRANSPORT / WNT-SIGNALLING | ||||||
| Function / homology |  Function and homology information Function and homology informationXAV939 stabilizes AXIN / positive regulation of telomere capping / NAD+ ADP-ribosyltransferase / negative regulation of telomere maintenance via telomere lengthening / protein auto-ADP-ribosylation / protein localization to chromosome, telomeric region / NAD+-protein-aspartate ADP-ribosyltransferase activity / protein poly-ADP-ribosylation / NAD+-protein-glutamate ADP-ribosyltransferase activity / NAD+-protein mono-ADP-ribosyltransferase activity ...XAV939 stabilizes AXIN / positive regulation of telomere capping / NAD+ ADP-ribosyltransferase / negative regulation of telomere maintenance via telomere lengthening / protein auto-ADP-ribosylation / protein localization to chromosome, telomeric region / NAD+-protein-aspartate ADP-ribosyltransferase activity / protein poly-ADP-ribosylation / NAD+-protein-glutamate ADP-ribosyltransferase activity / NAD+-protein mono-ADP-ribosyltransferase activity / pericentriolar material / NAD+ poly-ADP-ribosyltransferase activity / Transferases; Glycosyltransferases; Pentosyltransferases / positive regulation of telomere maintenance via telomerase / nucleotidyltransferase activity / TCF dependent signaling in response to WNT / Degradation of AXIN / Wnt signaling pathway / Regulation of PTEN stability and activity / protein polyubiquitination / positive regulation of canonical Wnt signaling pathway / nuclear envelope / chromosome, telomeric region / Ub-specific processing proteases / Golgi membrane / perinuclear region of cytoplasm / enzyme binding / metal ion binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Karlberg, T. / Schutz, P. / Arrowsmith, C.H. / Berglund, H. / Bountra, C. / Collins, R. / Edwards, A.M. / Flodin, S. / Flores, A. / Graslund, S. ...Karlberg, T. / Schutz, P. / Arrowsmith, C.H. / Berglund, H. / Bountra, C. / Collins, R. / Edwards, A.M. / Flodin, S. / Flores, A. / Graslund, S. / Hammarstrom, M. / Johansson, I. / Kotenyova, T. / Markova, N. / Moche, M. / Nordlund, P. / Nyman, T. / Persson, C. / Siponen, M.I. / Svensson, L. / Thorsell, A.G. / Tresaugues, L. / Van Den Berg, S. / Weigelt, J. / Welin, M. / Wisniewska, M. / Schuler, H. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: Nat.Biotechnol. / Year: 2012 Title: Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors Authors: Wahlberg, E. / Karlberg, T. / Kouznetsova, E. / Markova, N. / Macchiarulo, A. / Thorsell, A.G. / Pol, E. / Frostell, A. / Ekblad, T. / Kull, B. / Robertson, G.M. / Pellicciari, R. / Schuler, H. / Weigelt, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3mhj.cif.gz | 109.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3mhj.ent.gz | 82.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3mhj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3mhj_validation.pdf.gz | 1 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3mhj_full_validation.pdf.gz | 1 MB | Display | |
| Data in XML | 3mhj_validation.xml.gz | 22.1 KB | Display | |
| Data in CIF | 3mhj_validation.cif.gz | 31.6 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/mh/3mhjftp://data.pdbj.org/pub/pdb/validation_reports/mh/3mhj | HTTPS FTP |
-Related structure data
| Related structure data |  3goyC  3mhkC  3p0nC  3p0pC  3p0qC  3se2C  3smiC  3smjC  3kr7S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 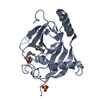
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27299.764 Da / Num. of mol.: 2 / Fragment: C-TERMINAL PARP DOMAIN (UNP RESIDUES 946-1162) Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PARP5B, TANK2, TNKL, TNKS2 / Plasmid: PNIC-BSA4 / Production host:  #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | 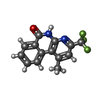  Mass: 278.229 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H9F3N2O Mass: 278.229 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H9F3N2O#4: Chemical |   Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 343 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 343 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.41 Å3/Da / Density % sol: 49.03 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: 17% PEG 3350, 0.2M Ammonium Sulfate, 0.1M Tris-HCl, pH 7.5, VAPOR DIFFUSION, SITTING DROP, temperature 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID29 / Wavelength: 0.97908 Å / Beamline: ID29 / Wavelength: 0.97908 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Dec 17, 2009 Details: liquid nitrogen cooled channel-cut silicon monochromator and cylindrical grazing incidence mirror |
| Radiation | Monochromator: Si monochromator crystals / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97908 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→45.21 Å / Num. all: 49107 / Num. obs: 49107 / % possible obs: 99.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 7.1 % / Rmerge(I) obs: 0.054 / Rsym value: 0.057 / Net I/σ(I): 24.6 |
| Reflection shell | Resolution: 1.8→1.85 Å / Redundancy: 5 % / Rmerge(I) obs: 0.452 / Mean I/σ(I) obs: 3.7 / Num. unique all: 3582 / Rsym value: 0.429 / % possible all: 99.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3KR7 Resolution: 1.8→45.21 Å / Cor.coef. Fo:Fc: 0.959 / Cor.coef. Fo:Fc free: 0.941 / SU B: 2.062 / SU ML: 0.066 / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.109 / ESU R Free: 0.106 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.601 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→45.21 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.847 Å / Total num. of bins used: 20
|
