 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
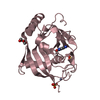
Yorodumi- PDB-4uvp: Crystal structure of human tankyrase 2 in complex with 5-amino-3-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 4uvp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human tankyrase 2 in complex with 5-amino-3- ethyl-1,2-dihydroisoquinolin-1-one | ||||||
 Components Components | (TANKYRASE-2) x 2 | ||||||
 Keywords Keywords | TRANSFERASE / PROTEIN-LIGAND COMPLEX / DIPHTHERIA TOXIN LIKE FOLD / ADP-RIBOSYLATION / TRANSFERASE-TRANSFERASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationXAV939 stabilizes AXIN / positive regulation of telomere capping / NAD+ ADP-ribosyltransferase / protein auto-ADP-ribosylation / negative regulation of telomere maintenance via telomere lengthening / protein localization to chromosome, telomeric region / NAD+-protein-aspartate ADP-ribosyltransferase activity / protein poly-ADP-ribosylation / NAD+-protein-glutamate ADP-ribosyltransferase activity / NAD+-protein mono-ADP-ribosyltransferase activity ...XAV939 stabilizes AXIN / positive regulation of telomere capping / NAD+ ADP-ribosyltransferase / protein auto-ADP-ribosylation / negative regulation of telomere maintenance via telomere lengthening / protein localization to chromosome, telomeric region / NAD+-protein-aspartate ADP-ribosyltransferase activity / protein poly-ADP-ribosylation / NAD+-protein-glutamate ADP-ribosyltransferase activity / NAD+-protein mono-ADP-ribosyltransferase activity / pericentriolar material / Transferases; Glycosyltransferases; Pentosyltransferases / NAD+ poly-ADP-ribosyltransferase activity / positive regulation of telomere maintenance via telomerase / nucleotidyltransferase activity / TCF dependent signaling in response to WNT / Degradation of AXIN / Wnt signaling pathway / Regulation of PTEN stability and activity / protein polyubiquitination / positive regulation of canonical Wnt signaling pathway / nuclear envelope / chromosome, telomeric region / Ub-specific processing proteases / Golgi membrane / perinuclear region of cytoplasm / enzyme binding / metal ion binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å | ||||||
 Authors Authors | Narwal, M. / Haikarainen, T. / Lehtio, L. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem. / Year: 2015 Title: Exploration of the Nicotinamide-Binding Site of the Tankyrases, Identifying 3-Arylisoquinolin-1-Ones as Potent and Selective Inhibitors in Vitro. Authors: Paine, H.A. / Nathubhai, A. / Woon, E.C.Y. / Sunderland, P.T. / Wood, P.J. / Mahon, M.F. / Lloyd, M.D. / Thompson, A.S. / Haikarainen, T. / Narwal, M. / Lehtio, L. / Threadgill, M.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 4uvp.cif.gz | 111 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb4uvp.ent.gz | 85.9 KB | Display | PDB format |
| PDBx/mmJSON format | 4uvp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/uv/4uvpftp://data.pdbj.org/pub/pdb/validation_reports/uv/4uvp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4uvlC  4uvnC  4uvoC  4uvsC  4uvtC  4uvuC  4uvvC  4uvwC  4uvxC  4uvyC  4uvzC  3kr7S C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| 2 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
|
-Components
-Protein / Protein/peptide , 2 types, 4 molecules ACBD
| #1: Protein | Mass: 21824.545 Da / Num. of mol.: 2 / Fragment: C-TERMINAL FRAGMENT, RESIDUES 946-1113 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PNIC28-BSA4 / Production host:  #2: Protein/peptide | Mass: 5364.037 Da / Num. of mol.: 2 / Fragment: C-TERMINAL FRAGMENT, RESIDUES 1115-1162 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Plasmid: PNIC28-BSA4 / Production host: |
|---|
-Non-polymers , 6 types, 462 molecules 


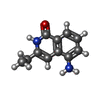


| #3: Chemical |  Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4#5: Chemical |  Mass: 106.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H10O3#6: Chemical |  Mass: 188.226 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H12N2O Mass: 188.226 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C11H12N2O#7: Chemical | ChemComp-GOL / |  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 451 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Sequence details | GAP IN THE PROTEIN CHAIN DUE TO CHYMOTRYPS |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 49 % / Description: NONE |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 8.5 Details: 0.2 M LISO4, 0.1 M TRIS HCL, 22% PEG3350, PH 8.5, VAPOR DIFFUSION, SITTING DROP, TEMPERATURE 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-1 / Wavelength: 1.07227 / Beamline: ID23-1 / Wavelength: 1.07227 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Apr 8, 2011 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.07227 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→50 Å / Num. obs: 52759 / % possible obs: 98.4 % / Observed criterion σ(I): -3 / Redundancy: 3.5 % / Rmerge(I) obs: 0.05 / Net I/σ(I): 16.29 |
| Reflection shell | Resolution: 1.75→1.8 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.46 / Mean I/σ(I) obs: 2.19 / % possible all: 85.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3KR7 Resolution: 1.75→45.17 Å / Cor.coef. Fo:Fc: 0.962 / Cor.coef. Fo:Fc free: 0.943 / SU B: 2.01 / SU ML: 0.065 / Cross valid method: THROUGHOUT / ESU R: 0.101 / ESU R Free: 0.102 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 17.446 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→45.17 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|