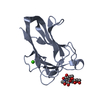
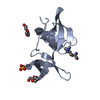
- PDB-3lgl: Crystal structure of the 53BP1 tandem tudor domain in complex wit... -
+
データを開く
IDまたはキーワード:
読み込み中...
-
基本情報
登録情報
データベース: PDB / ID: 3lgl
タイトル
Crystal structure of the 53BP1 tandem tudor domain in complex with p53K382me2
要素
DIMETHYLATED p53 LYSINE 382 PEPTIDE
Tumor suppressor p53-binding protein 1
キーワード
CELL CYCLE / TANDEM TUDOR DOMAIN / DIMETHYLATED p53 PEPTIDE / DNA REPAIR / DNA damage / DNA-binding / Methylation / Transcription / Transcription regulation
機能・相同性
機能・相同性情報
ubiquitin-modified histone reader activity / positive regulation of isotype switching / histone H4K20me2 reader activity / cellular response to X-ray / double-strand break repair via classical nonhomologous end joining / protein localization to site of double-strand break / DNA repair complex / positive regulation of intrinsic apoptotic signaling pathway by p53 class mediator / telomeric DNA binding / : ...ubiquitin-modified histone reader activity / positive regulation of isotype switching / histone H4K20me2 reader activity / cellular response to X-ray / double-strand break repair via classical nonhomologous end joining / protein localization to site of double-strand break / DNA repair complex / positive regulation of intrinsic apoptotic signaling pathway by p53 class mediator / telomeric DNA binding / : / histone reader activity / SUMOylation of transcription factors / negative regulation of double-strand break repair via homologous recombination / DNA damage checkpoint signaling / replication fork / transcription coregulator activity / Nonhomologous End-Joining (NHEJ) / G2/M DNA damage checkpoint / protein homooligomerization / kinetochore / double-strand break repair via nonhomologous end joining / p53 binding / Recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double strand breaks / site of double-strand break / Processing of DNA double-strand break ends / histone binding / damaged DNA binding / RNA polymerase II-specific DNA-binding transcription factor binding / transcription coactivator activity / chromosome, telomeric region / nuclear body / DNA damage response / positive regulation of DNA-templated transcription / positive regulation of transcription by RNA polymerase II / nucleoplasm / nucleus / cytoplasm 類似検索 - 分子機能
Tumour suppressor p53-binding protein-1 Tudor domain / : / Tumour suppressor p53-binding protein-1 Tudor / BRCA1 C Terminus (BRCT) domain / : / : / SH3 type barrels. - #30 / SH3 type barrels. - #140 / breast cancer carboxy-terminal domain / BRCT domain profile. ...Tumour suppressor p53-binding protein-1 Tudor domain / : / Tumour suppressor p53-binding protein-1 Tudor / BRCA1 C Terminus (BRCT) domain / : / : / SH3 type barrels. - #30 / SH3 type barrels. - #140 / breast cancer carboxy-terminal domain / BRCT domain profile. / BRCT domain / BRCT domain superfamily / SH3 type barrels. / Ribosomal protein L2, domain 2 / Roll / Mainly Beta 類似検索 - ドメイン・相同性
TRIETHYLENE GLYCOL / TP53-binding protein 1 類似検索 - 構成要素
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Homo sapiens (ヒト)
Homo sapiens (ヒト) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード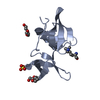










 PDBj
PDBj







 集合体
集合体


 分子量: 150.173 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C6H14O4
分子量: 150.173 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C6H14O4
 分子量: 96.063 Da / 分子数: 1 / 由来タイプ: 合成 / 式: SO4
分子量: 96.063 Da / 分子数: 1 / 由来タイプ: 合成 / 式: SO4 分子量: 18.015 Da / 分子数: 125 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 125 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 4.2.2 / 波長: 1 Å
/ ビームライン: 4.2.2 / 波長: 1 Å 解析
解析