 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3l36 | ||||||
|---|---|---|---|---|---|---|---|
| Title | PIE12 D-peptide against HIV entry | ||||||
 Components Components |
| ||||||
 Keywords Keywords | DE NOVO PROTEIN / COILED-COIL / D-PEPTIDE INHIBITOR | ||||||
| Function / homology | Single alpha-helices involved in coiled-coils or other helix-helix interfaces - #170 / Single alpha-helices involved in coiled-coils or other helix-helix interfaces / Up-down Bundle / Mainly Alpha Function and homology information Function and homology information | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.45 Å | ||||||
 Authors Authors | Welch, B.D. / Redman, J.S. / Paul, S. / Whitby, F.G. / Weinstock, M.T. / Reeves, J.D. / Lie, Y.S. / Eckert, D.M. / Hill, C.P. / Root, M.J. / Kay, M.S. | ||||||
 Citation Citation | Journal: J.Virol. / Year: 2010 Title: Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. Authors: Welch, B.D. / Francis, J.N. / Redman, J.S. / Paul, S. / Weinstock, M.T. / Reeves, J.D. / Lie, Y.S. / Whitby, F.G. / Eckert, D.M. / Hill, C.P. / Root, M.J. / Kay, M.S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3l36.cif.gz | 23.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3l36.ent.gz | 19.1 KB | Display | PDB format |
| PDBx/mmJSON format | 3l36.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3l36_validation.pdf.gz | 448.4 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3l36_full_validation.pdf.gz | 448.5 KB | Display | |
| Data in XML | 3l36_validation.xml.gz | 5.3 KB | Display | |
| Data in CIF | 3l36_validation.cif.gz | 6.7 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/l3/3l36ftp://data.pdbj.org/pub/pdb/validation_reports/l3/3l36 | HTTPS FTP |
-Related structure data













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||
| Unit cell |
| |||||||||
| Components on special symmetry positions |
| |||||||||
| Details | BIOMOLECULE: 1, 2 SEE REMARK 350 FOR THE AUTHOR PROVIDED AND/OR PROGRAM GENERATED ASSEMBLY INFORMATION FOR THE STRUCTURE IN THIS ENTRY. THE REMARK MAY ALSO PROVIDE INFORMATION ON BURIED SURFACE AREA. COORDINATES FOR A COMPLETE MULTIMER REPRESENTING THE KNOWN BIOLOGICALLY SIGNIFICANT OLIGOMERIZATION STATE OF THE MOLECULE CAN BE GENERATED BY APPLYING BIOMT TRANSFORMATIONS GIVEN BELOW. BOTH NON-CRYSTALLOGRAPHIC AND CRYSTALLOGRAPHIC OPERATIONS ARE GIVEN. BIOMOLECULE: 1 AUTHOR DETERMINED BIOLOGICAL UNIT: HEXAMERIC SOFTWARE DETERMINED QUATERNARY STRUCTURE: HEXAMERIC SOFTWARE USED: PISA TOTAL BURIED SURFACE AREA: 12270 ANGSTROM**2 SURFACE AREA OF THE COMPLEX: 10770 ANGSTROM**2 CHANGE IN SOLVENT FREE ENERGY: -110.0 KCAL/MOL APPLY THE FOLLOWING TO CHAINS: A, H BIOMT1 1 1.000000 0.000000 0.000000 0.00000 BIOMT2 1 0.000000 1.000000 0.000000 0.00000 BIOMT3 1 0.000000 0.000000 1.000000 0.00000 BIOMT1 2 -0.500000 -0.866025 0.000000 20.55850 BIOMT2 2 0.866025 -0.500000 0.000000 -35.60837 BIOMT3 2 0.000000 0.000000 1.000000 0.00000 BIOMT1 3 -0.500000 0.866025 0.000000 41.11700 BIOMT2 3 -0.866025 -0.500000 0.000000 0.00000 BIOMT3 3 0.000000 0.000000 1.000000 0.00000 BIOMOLECULE: 2 SOFTWARE DETERMINED QUATERNARY STRUCTURE: DIMERIC SOFTWARE USED: PISA TOTAL BURIED SURFACE AREA: 1570 ANGSTROM**2 SURFACE AREA OF THE COMPLEX: 6110 ANGSTROM**2 CHANGE IN SOLVENT FREE ENERGY: -16.0 KCAL/MOL APPLY THE FOLLOWING TO CHAINS: A, H BIOMT1 1 1.000000 0.000000 0.000000 0.00000 BIOMT2 1 0.000000 1.000000 0.000000 0.00000 BIOMT3 1 0.000000 0.000000 1.000000 0.00000 |
-Components
| #1: Protein/peptide | Mass: 5466.574 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: L-PEPTIDE WITH N-TERMINAL ACETYL GROUP AND C-TERMINAL AMIDE GROUP |
|---|---|
| #2: Protein/peptide | Mass: 1972.249 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: D-PEPTIDE WITH N-TERMINAL ACETYL GROUP AND C-TERMINAL AMIDE GROUP |
| #3: Chemical | ChemComp-CXS / 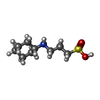  Mass: 221.317 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H19NO3S / Comment: pH buffer*YM Mass: 221.317 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C9H19NO3S / Comment: pH buffer*YM |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.68 Å3/Da / Density % sol: 54.12 % |
|---|---|
| Crystal grow | pH: 10.5 Details: 0.1 M CAPS PH 10.5, 40% MPD, VAPOR DIFFUSION, SITTING DROP, TEMPERATURE 277K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL11-1 / Wavelength: 0.97945 / Beamline: BL11-1 / Wavelength: 0.97945 |
| Detector | Type: MARMOSAIC 325 mm CCD / Detector: CCD / Date: Jun 11, 2008 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97945 Å / Relative weight: 1 |
| Reflection | Resolution: 1.45→30 Å / Num. obs: 14802 / % possible obs: 99.7 % / Observed criterion σ(I): 0 / Redundancy: 12.6 % / Rmerge(I) obs: 0.107 / Net I/σ(I): 22.644 |
| Reflection shell | Resolution: 1.45→1.5 Å / Redundancy: 6.6 % / Rmerge(I) obs: 0.235 / % possible all: 96.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.45→21.64 Å / Cor.coef. Fo:Fc: 0.947 / Cor.coef. Fo:Fc free: 0.92 / SU B: 1.122 / SU ML: 0.045 / Cross valid method: THROUGHOUT / ESU R: 0.083 / ESU R Free: 0.086 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES: REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 29.18 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.45→21.64 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.45→1.49 Å / Total num. of bins used: 20
|
