 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3ker: D-Dopachrome tautomerase (D-DT)/ macrophage migration inhibitory ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3ker | ||||||
|---|---|---|---|---|---|---|---|
| Title | D-Dopachrome tautomerase (D-DT)/ macrophage migration inhibitory factor 2 (MIF2) complexed with inhibitor 4-IPP | ||||||
 Components Components | D-dopachrome decarboxylase | ||||||
 Keywords Keywords | CYTOKINE/INHIBITOR / Tautomerase / Inflammation / Cytokine / CYTOKINE-INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationD-dopachrome decarboxylase / D-dopachrome decarboxylase activity / melanin biosynthetic process / phenylpyruvate tautomerase activity / cytokine receptor binding / positive regulation of inflammatory response / protease binding / : / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.78 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.78 Å | ||||||
 Authors Authors | Zierow, S. / Lolis, E. | ||||||
 Citation Citation | Journal: Faseb J. / Year: 2014 Title: Targeting distinct tautomerase sites of D-DT and MIF with a single molecule for inhibition of neutrophil lung recruitment. Authors: Rajasekaran, D. / Zierow, S. / Syed, M. / Bucala, R. / Bhandari, V. / Lolis, E.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3ker.cif.gz | 102.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3ker.ent.gz | 80.5 KB | Display | PDB format |
| PDBx/mmJSON format | 3ker.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ke/3kerftp://data.pdbj.org/pub/pdb/validation_reports/ke/3ker | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3kanSC  4q3fC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain: (Details: chain A,A,A, using strict) NCS oper:
|
-Components
| #1: Protein | Mass: 12959.976 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | ChemComp-RW1 / 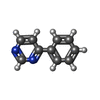  Mass: 156.184 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H8N2 Mass: 156.184 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H8N2#3: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: Cl#4: Chemical | ChemComp-NA /   Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Na#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 8 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 8 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.71 Å3/Da / Density % sol: 54.65 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 1M Sodium Citrate, 0.1M Tris, 0.2M NaCl, pH 7, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Oct 15, 2009 / Details: mirrors |
| Radiation | Monochromator: Graphite / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.78→50 Å / Num. obs: 13925 / % possible obs: 99.8 % / Redundancy: 4.3 % / Rmerge(I) obs: 0.121 / Χ2: 0.732 / Net I/σ(I): 6.2 |
| Reflection shell | Resolution: 2.78→2.88 Å / Redundancy: 4.2 % / Rmerge(I) obs: 0.514 / Num. unique all: 1384 / Χ2: 0.63 / % possible all: 100 |
-Phasing
| Phasing | Method: molecular replacement | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MR | Rfactor: 33.07 / Model details: Phaser MODE: MR_AUTO
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3KAN Resolution: 2.78→31.23 Å / Occupancy max: 1 / Occupancy min: 1 / FOM work R set: 0.809 / Cross valid method: THROUGHOUT / σ(F): 0 Details: One protein chain was refined using strict NCS, and NCS symmetry operators were used to generate the other three peptide polymers in the asymmetric unit. NCS averaged maps were also used. ...Details: One protein chain was refined using strict NCS, and NCS symmetry operators were used to generate the other three peptide polymers in the asymmetric unit. NCS averaged maps were also used. The deposited model therefore is an averaged model from all four peptide polymers in the asymmetric unit. The final model deposited has been generated by real-space transformations of chain A. The real space rotation matrix to generate chain B is described by: ( -0.24621 0.22510 0.94271 ) ( 0.82241 0.56320 0.08031 ) ( -0.51286 0.79507 -0.32379 ). The real space translation vector is described by: (4.46522 -21.53777 46.77695). Chain C was generated using real space rotation matrix: ( -0.23956 0.81293 -0.53080 ) ( 0.23408 0.57896 0.78104 ) ( 0.94224 0.06285 -0.32898 ) and real-space translation vector: (42.49111 -25.63348 12.28672). Chain D was generated using real space rotation matrix: ( -0.23956 0.81293 -0.53080 ) ( 0.23408 0.57896 0.78104 ) ( 0.94224 0.06285 -0.32898) and the real-space translation vector: (47.24985 5.40733 42.11218)
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Bsol: 42.23 Å2 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 79.24 Å2 / Biso mean: 42.715 Å2 / Biso min: 23.36 Å2
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.78→31.23 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS |
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
|
