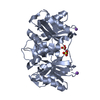
Entry Database : PDB / ID : 3k7vTitle Protein phosphatase 2A core complex bound to dinophysistoxin-1 (Serine/threonine-protein phosphatase 2A ...) x 2 Keywords / / / / / / / / / / / / / / / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / Resolution : 2.85 Å Authors Jeffrey, P.D. / Huhn, J. / Shi, Y. Journal : Chem.Res.Toxicol. / Year : 2009Title : A structural basis for the reduced toxicity of dinophysistoxin-2.Authors : Huhn, J. / Jeffrey, P.D. / Larsen, K. / Rundberget, T. / Rise, F. / Cox, N.R. / Arcus, V. / Shi, Y. / Miles, C.O. History Deposition Oct 13, 2009 Deposition site / Processing site Revision 1.0 Nov 3, 2009 Provider / Type Revision 1.1 Jul 13, 2011 Group Revision 1.2 Sep 6, 2023 Group Data collection / Database references ... Data collection / Database references / Derived calculations / Refinement description Category chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model / pdbx_struct_conn_angle / struct_conn / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr2_auth_seq_id / _pdbx_struct_conn_angle.ptnr2_label_asym_id / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id
Show all Show less
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads







 PDBj
PDBj

















 Assembly
Assembly


 Spodoptera frugiperda (fall armyworm) / Strain (production host): HI-5
Spodoptera frugiperda (fall armyworm) / Strain (production host): HI-5


 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4
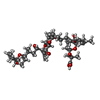
Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 819.029 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C45H70O13
Mass: 819.029 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C45H70O13 Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn
Mass: 54.938 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mn Sample preparation
Sample preparation / Beamline: X29A / Wavelength: 1.0809 Å
/ Beamline: X29A / Wavelength: 1.0809 Å Processing
Processing