 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3hm8: Crystal structure of the C-terminal Hexokinase domain of human HK3 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3hm8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the C-terminal Hexokinase domain of human HK3 | ||||||
 Components Components | Hexokinase-3 | ||||||
 Keywords Keywords | TRANSFERASE / glucose / glucose-6-phosphate / non-protein kinase / hexokinase / STRUCTURAL GENOMICS / STRUCTURAL GENOMICS CONSORTIUM / SGC / Allosteric enzyme / ATP-binding / Glycolysis / Kinase / Nucleotide-binding | ||||||
| Function / homology |  Function and homology information Function and homology informationhexokinase activity / mannokinase activity / hexokinase / fructokinase activity / glucokinase activity / glucose 6-phosphate metabolic process / D-glucose binding / fructose 6-phosphate metabolic process / Glycolysis / canonical glycolysis ...hexokinase activity / mannokinase activity / hexokinase / fructokinase activity / glucokinase activity / glucose 6-phosphate metabolic process / D-glucose binding / fructose 6-phosphate metabolic process / Glycolysis / canonical glycolysis / intracellular glucose homeostasis / glycolytic process / glucose metabolic process / secretory granule lumen / ficolin-1-rich granule lumen / Neutrophil degranulation / mitochondrion / extracellular region / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / molecular replacement / Resolution: 2.8 Å X-RAY DIFFRACTION / SYNCHROTRON / molecular replacement / Resolution: 2.8 Å | ||||||
 Authors Authors | Nedyalkova, L. / Tong, Y. / Rabeh, W. / Tempel, W. / Landry, R. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Weigelt, J. / Bochkarev, A. ...Nedyalkova, L. / Tong, Y. / Rabeh, W. / Tempel, W. / Landry, R. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Weigelt, J. / Bochkarev, A. / Park, H. / Structural Genomics Consortium (SGC) | ||||||
 Citation Citation | Journal: To be Published Title: Crystal structure of the C-terminal Hexokinase domain of human HK3 Authors: Nedyalkova, L. / Tong, Y. / Rabeh, W. / Tempel, W. / Landry, R. / Arrowsmith, C.H. / Edwards, A.M. / Bountra, C. / Weigelt, J. / Bochkarev, A. / Park, H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3hm8.cif.gz | 642.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3hm8.ent.gz | 534.2 KB | Display | PDB format |
| PDBx/mmJSON format | 3hm8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/hm/3hm8ftp://data.pdbj.org/pub/pdb/validation_reports/hm/3hm8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2nztS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS ensembles :
|
-Components
| #1: Protein | Mass: 48368.410 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: HK3 / Plasmid: pET28a-LIC / Production host:  #2: Sugar | ChemComp-GLC / 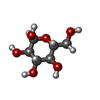  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 4 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C6H12O6 #3: Sugar | ChemComp-BG6 / 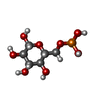  Type: D-saccharide, beta linking / Mass: 260.136 Da / Num. of mol.: 4 Type: D-saccharide, beta linking / Mass: 260.136 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C6H13O9P |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3 Å3/Da / Density % sol: 58.4 % |
|---|---|
| Crystal grow | Temperature: 291 K / pH: 7.2 Details: reservoir solution: 16% PEG 3350, 0.20 M Ammonium Citrate, 0.1 M HEPES. 0.005 M glucose-6-phosphate, 0.005 M ADP-Mg, 0.02 M TCEP, 1:100 (w/w) trypsin was added to the protein stock solution. ...Details: reservoir solution: 16% PEG 3350, 0.20 M Ammonium Citrate, 0.1 M HEPES. 0.005 M glucose-6-phosphate, 0.005 M ADP-Mg, 0.02 M TCEP, 1:100 (w/w) trypsin was added to the protein stock solution., pH 7.2, vapor diffusion, sitting drop, temperature 291K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 19-ID / Wavelength: 0.97857 / Beamline: 19-ID / Wavelength: 0.97857 |
| Detector | Type: ADSC QUANTUM 315 / Detector: CCD / Date: Mar 20, 2009 |
| Radiation | Protocol: SINGLE WAVELENGTH / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97857 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→20 Å / Num. obs: 53686 / % possible obs: 98.4 % |
| Reflection shell | Resolution: 2.8→2.9 Å / Rmerge(I) obs: 0.666 / % possible all: 86.8 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: molecular replacement Starting model: pdb entry 2NZT, chain A, residues 468 through 913 Resolution: 2.8→20 Å / Cor.coef. Fo:Fc: 0.938 / Cor.coef. Fo:Fc free: 0.915 / Occupancy max: 1 / Occupancy min: 1 / SU B: 48.382 / SU ML: 0.384 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE PROGRAMS CHAINSAW, PRODRG, PHENIX, COOT AND MOLPROBLITY WERE ALSO USED.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 69.23 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→20 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | Refine-ID: X-RAY DIFFRACTION
|