 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3gfq | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of YhdA, K109L variant | ||||||
 Components Components | FMN-dependent NADPH-azoreductase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / FLAVOPROTEINS / QUINONE REDUCTASE / FLAVODOXIN / OLIGOMERIZATION / Flavoprotein / FMN / NADP | ||||||
| Function / homology |  Function and homology information Function and homology informationOxidoreductases; Acting on other nitrogenous compounds as donors / FMN binding / oxidoreductase activity / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.996 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.996 Å | ||||||
 Authors Authors | Staunig, N. / Gruber, K. | ||||||
 Citation Citation | Journal: Febs J. / Year: 2009 Title: A single intersubunit salt bridge affects oligomerization and catalytic activity in a bacterial quinone reductase Authors: Binter, A. / Staunig, N. / Jelesarov, I. / Lohner, K. / Palfey, B.A. / Deller, S. / Gruber, K. / Macheroux, P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3gfq.cif.gz | 125.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3gfq.ent.gz | 100.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3gfq.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gf/3gfqftp://data.pdbj.org/pub/pdb/validation_reports/gf/3gfq | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3gfrC  3gfsC  1nniS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Ens-ID: 1 / End auth comp-ID: ALA / End label comp-ID: ALA
|
-Components
| #1: Protein | Mass: 18909.848 Da / Num. of mol.: 4 / Mutation: K109L Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: O07529, Oxidoreductases; Acting on other nitrogenous compounds as donors #2: Chemical | ChemComp-FMN / 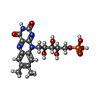  Mass: 456.344 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C17H21N4O9P Mass: 456.344 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C17H21N4O9P |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46.47 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: batch crystallization / pH: 7.5 Details: 0.2 M Ammonium Sulfate, 0.1 M HEPES pH 7.5, 25% w/v PEG 3350, batch crystallization, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.801 Å / Beamline: X11 / Wavelength: 0.801 Å |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Jul 12, 2005 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.801 Å / Relative weight: 1 |
| Reflection | Resolution: 2.996→29.1 Å / Num. all: 14641 / Num. obs: 14641 / % possible obs: 99.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.8 % / Biso Wilson estimate: 59.61 Å2 / Rsym value: 0.179 |
| Reflection shell | Resolution: 2.996→3.05 Å / Redundancy: 4.5 % / Mean I/σ(I) obs: 1.5 / Rsym value: 0.908 / % possible all: 99.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 1nni Resolution: 2.996→29.097 Å / Occupancy max: 1 / Occupancy min: 1 / FOM work R set: 0.797 / SU ML: 0.45 Isotropic thermal model: a single isotropic temperature factor per residue Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 41.2 Å2 / ksol: 0.346 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 176.88 Å2 / Biso mean: 59.551 Å2 / Biso min: 24.94 Å2
| ||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.996→29.097 Å
| ||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS |
| ||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 5
|
