 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3fxu: Crystal structure of TsaR in complex with its effector p-toluenes... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3fxu | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of TsaR in complex with its effector p-toluenesulfonate | ||||||
 Components Components | LysR type regulator of tsaMBCD | ||||||
 Keywords Keywords | Transcription regulator / LysR-type / transcriptional regulator / LTTR / TsaR / wHTH / DNA-binding / Transcription / Transcription regulation | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Comamonas testosteroni (bacteria) Comamonas testosteroni (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.95 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.95 Å | ||||||
 Authors Authors | Monferrer, D. / Tralau, T. / Kertesz, M.A. / Kikhney, A. / Svergun, D. / Uson, I. | ||||||
 Citation Citation | Journal: Mol.Microbiol. / Year: 2010 Title: Structural studies on the full-length LysR-type regulator TsaR from Comamonas testosteroni T-2 reveal a novel open conformation of the tetrameric LTTR fold Authors: Monferrer, D. / Tralau, T. / Kertesz, M.A. / Dix, I. / Sola, M. / Uson, I. #1: Journal: Acta Crystallogr.,Sect.F / Year: 2008 Title: High crystallizability under air-exclusion conditions of the full-length LysR-type transcriptional regulator TsaR from Comamonas testosteroni T-2 and data-set analysis for a MIRAS structure-solution approach Authors: Monferrer, D. / Tralau, T. / Kertesz, M.A. / Panjikar, S. / Uson, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3fxu.cif.gz | 130.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3fxu.ent.gz | 102.4 KB | Display | PDB format |
| PDBx/mmJSON format | 3fxu.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fx/3fxuftp://data.pdbj.org/pub/pdb/validation_reports/fx/3fxu | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3fxqSC  3fxrC  3fzjC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj






- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 33643.727 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Comamonas testosteroni (bacteria) / Strain: T-2 / Gene: tsaR / Plasmid: PQE-70 / Production host: |
|---|
-Non-polymers , 6 types, 227 molecules 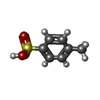





| #2: Chemical | ChemComp-TSU /  Mass: 172.202 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: C7H8O3S Mass: 172.202 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: C7H8O3S#3: Chemical | ChemComp-IMD / |  Mass: 69.085 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H5N2 Mass: 69.085 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H5N2#4: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H8O3#5: Chemical |  Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Cl#6: Chemical | ChemComp-FMT / |  Mass: 46.025 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH2O2 Mass: 46.025 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: CH2O2#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 207 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Sequence details | THE SEQUENCE OF DATABASE UNIPROTKB/TREMBL P94678 (P94678_COMTE) HAS A SEQUENCING ERROR.THE ...THE SEQUENCE OF DATABASE UNIPROTKB/TREMBL P94678 (P94678_COMTE) HAS A SEQUENCING |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.66 Å3/Da / Density % sol: 53.79 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: microbatch under oil / pH: 4.6 Details: 2M Na formate, 0.1M Na acetate pH 4.6. Soaking in 250mM p-toluenesulfonate, microbatch under oil, temperature 298K |
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.9326 Å / Beamline: ID14-2 / Wavelength: 0.9326 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Nov 29, 2007 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9326 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→19.75 Å / Num. all: 56186 / Num. obs: 55862 / % possible obs: 99.4 % / Redundancy: 0.99 % / Rmerge(I) obs: 0.0607 / Rsym value: 0.0453 / Net I/σ(I): 17.95 |
| Reflection shell | Resolution: 1.9→1.99 Å / Rmerge(I) obs: 0.4144 / Mean I/σ(I) obs: 2.62 / Num. unique all: 7242 / Rsym value: 0.4405 / % possible all: 97 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB entry 3FXQ Resolution: 1.95→19.75 Å / Cor.coef. Fo:Fc: 0.932 / Cor.coef. Fo:Fc free: 0.908 / Occupancy max: 1 / Occupancy min: 0.5 / SU B: 9.103 / SU ML: 0.119 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R: 0.176 / ESU R Free: 0.159 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: U VALUES: RESIDUAL ONLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 62.3 Å2 / Biso mean: 20.663 Å2 / Biso min: 2 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.95→19.75 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.95→2.001 Å / Total num. of bins used: 20
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
