 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3fxe: Crystal structure of interacting domains of IcmR and IcmQ (seleno... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3fxe | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of interacting domains of IcmR and IcmQ (seleno-derivative) | ||||||
 Components Components |
| ||||||
 Keywords Keywords | UNKNOWN FUNCTION / 4 helix bundle / helix-turn-helix / Se-Met | ||||||
| Function / homology |  Function and homology information Function and homology information: / IcmR, middle region / Methane Monooxygenase Hydroxylase; Chain G, domain 1 - #90 / Immunoglobulin FC, subunit C / Single alpha-helices involved in coiled-coils or other helix-helix interfaces / Methane Monooxygenase Hydroxylase; Chain G, domain 1 / Up-down Bundle / Mainly Alpha Similarity search - Domain/homology | ||||||
| Biological species |   Legionella pneumophila (bacteria)Legionella pneumophila subsp. pneumophila str. Philadelphia 1 (bacteria) Legionella pneumophila (bacteria)Legionella pneumophila subsp. pneumophila str. Philadelphia 1 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.2 Å | ||||||
 Authors Authors | Raychaudhury, S. / Akey, C.W. / Head, J.F. | ||||||
 Citation Citation | Journal: Structure / Year: 2009 Title: Structure and Function of Interacting IcmR-IcmQ Domains from a Type IVb Secretion System in Legionella pneumophila. Authors: Raychaudhury, S. / Farelli, J.D. / Montminy, T.P. / Matthews, M. / Menetret, J.F. / Dumenil, G. / Roy, C.R. / Head, J.F. / Isberg, R.R. / Akey, C.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3fxe.cif.gz | 32.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3fxe.ent.gz | 22 KB | Display | PDB format |
| PDBx/mmJSON format | 3fxe.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fx/3fxeftp://data.pdbj.org/pub/pdb/validation_reports/fx/3fxe | HTTPS FTP |
|---|
-Related structure data

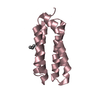





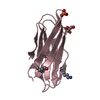



-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 6557.525 Da / Num. of mol.: 1 / Fragment: UNP residues 1-57 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Legionella pneumophila (bacteria) / Strain: Corby / Gene: icmQ, LPC_2899 / Plasmid: pET21b / Production host: |
|---|---|
| #2: Protein | Mass: 7859.662 Da / Num. of mol.: 1 / Fragment: UNP residues 23-95 / Mutation: L84M Source method: isolated from a genetically manipulated source Source: (gene. exp.) Legionella pneumophila subsp. pneumophila str. Philadelphia 1 (bacteria)Strain: Philadelphia-1 / DSM 7513 / Gene: icmR, lpg0443 / Plasmid: pET21b / Production host: |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 33 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.8 Å3/Da / Density % sol: 31.64 % |
|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 4.7 Details: 100 mM Na acetate pH 4.7, 30% PEG 1500, 100 mM L-cysteine, VAPOR DIFFUSION, HANGING DROP, temperature 295K |
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X12C / Wavelength: 0.97950, 0.97976, 0.95003 / Beamline: X12C / Wavelength: 0.97950, 0.97976, 0.95003 | ||||||||||||
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Aug 10, 2004 | ||||||||||||
| Radiation | Monochromator: Si(111) / Protocol: MAD / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||
| Radiation wavelength |
| ||||||||||||
| Reflection | Resolution: 2.2→50 Å / Num. all: 10087 / Num. obs: 10087 / % possible obs: 99.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 7.7 % / Rmerge(I) obs: 0.04 / Net I/σ(I): 23.9 | ||||||||||||
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 7.5 % / Rmerge(I) obs: 0.13 / Mean I/σ(I) obs: 18 / Num. unique all: 541 / % possible all: 99.4 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.2→50 Å / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→50 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
|
