 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3fl6: Influence of the incorporation of a cyclohexenyl nucleic acid (Ce... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3fl6 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Influence of the incorporation of a cyclohexenyl nucleic acid (CeNA) residue onto the sequence d(GCGTGCG)/d(CGCACGC) | ||||||
 Components Components |
| ||||||
 Keywords Keywords | DNA / double helix / CeNA / sugar modification / right-handed | ||||||
| Function / homology | COBALT HEXAMMINE(III) / DNA Function and homology information Function and homology information | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.17 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.17 Å | ||||||
 Authors Authors | Robeyns, K. / Herdewijn, P. / Van Meervelt, L. | ||||||
 Citation Citation | Journal: Artif DNA PNA XNA / Year: 2010 Title: Direct observation of two cyclohexenyl (CeNA) ring conformations in duplex DNA. Authors: Robeyns, K. / Herdewijn, P. / Van Meervelt, L. #1: Journal: Nucleic Acids Res. / Year: 2008Title: Influence of the incorporation of a cyclohexenyl nucleic acid (CeNA) residue onto the sequence d(CGCGAATTCGCG). Authors: Robeyns, K. / Herdewijn, P. / Van Meervelt, L. #2: Journal: J.Am.Chem.Soc. / Year: 2008Title: Structure of the fully modified left-handed cyclohexene nucleic acid sequence GTGTACAC. Authors: Robeyns, K. / Herdewijn, P. / Van Meervelt, L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3fl6.cif.gz | 47.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3fl6.ent.gz | 33.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3fl6.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 3fl6_validation.pdf.gz | 406.5 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 3fl6_full_validation.pdf.gz | 411.2 KB | Display | |
| Data in XML | 3fl6_validation.xml.gz | 6.1 KB | Display | |
| Data in CIF | 3fl6_validation.cif.gz | 7.9 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fl/3fl6ftp://data.pdbj.org/pub/pdb/validation_reports/fl/3fl6 | HTTPS FTP |
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |




-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 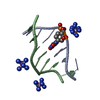
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 2164.460 Da / Num. of mol.: 2 / Source method: obtained synthetically #2: DNA chain | Mass: 2083.388 Da / Num. of mol.: 2 / Source method: obtained synthetically #3: Chemical | ChemComp-NCO / 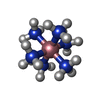  Mass: 161.116 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: CoH18N6 Mass: 161.116 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: CoH18N6#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 130 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.06 Å3/Da / Density % sol: 40.38 % | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 289 K / pH: 5.5 Details: 10%(v/v) 2-methyl-2,4-pentanediol (MPD), 20mM cobalt hexamine, 40mM potassium cacodylate buffered at pH=5.5, and 80/12mM KCl/NaCl, vapor diffusion, hanging drop, temperature 289K | ||||||||||||||||||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 0.7749 / Beamline: X06SA / Wavelength: 0.7749 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: PSI PILATUS 6M / Detector: PIXEL / Date: Apr 25, 2008 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: UNDULATOR / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.7749 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Redundancy: 8.8 % / Av σ(I) over netI: 2.1 / Number: 71625 / Rmerge(I) obs: 0.16 / Rsym value: 0.16 / D res high: 1.7 Å / D res low: 19.858 Å / Num. obs: 8126 / % possible obs: 98.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Diffraction reflection shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.17→19.86 Å / Num. obs: 24411 / % possible obs: 99.6 % / Observed criterion σ(I): 0 / Redundancy: 5.5 % / Biso Wilson estimate: 6.3 Å2 / Rmerge(I) obs: 0.094 / Rsym value: 0.094 / Net I/σ(I): 3.735 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Resolution: 1.17→1.23 Å / Redundancy: 5.9 % / Rmerge(I) obs: 0.52 / Mean I/σ(I) obs: 1.3 / Rsym value: 0.52 / % possible all: 98.8 |
-Phasing
| Phasing | Method: SAD | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phasing MAD set site |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 1.17→19.86 Å / Num. parameters: 6402 / Num. restraintsaints: 9922 / Occupancy max: 1 / Occupancy min: 0.22 / Cross valid method: FREE R / σ(F): 4 / Stereochemistry target values: ENGH AND HUBER Details: ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 4%
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201-228 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 10.739 Å2 | ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.17→19.86 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.17→1.22 Å /
|
