 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3eml: The 2.6 A Crystal Structure of a Human A2A Adenosine Receptor bou... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3eml | ||||||
|---|---|---|---|---|---|---|---|
| Title | The 2.6 A Crystal Structure of a Human A2A Adenosine Receptor bound to ZM241385. | ||||||
 Components Components | Human Adenosine A2A receptor/T4 lysozyme chimera | ||||||
 Keywords Keywords | MEMBRANE PROTEIN / RECEPTOR / adenosine / caffeine / GPCR / LCP / mesophase / Structural Genomics / PSI-2 / Protein Structure Initiative / Accelerated Technologies Center for Gene to 3D Structure / ATCG3D / GPCR Network | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of norepinephrine secretion / positive regulation of circadian sleep/wake cycle, sleep / Adenosine P1 receptors / positive regulation of acetylcholine secretion, neurotransmission / G protein-coupled adenosine receptor activity / response to purine-containing compound / G protein-coupled adenosine receptor signaling pathway / NGF-independant TRKA activation / Surfactant metabolism / type 5 metabotropic glutamate receptor binding ...regulation of norepinephrine secretion / positive regulation of circadian sleep/wake cycle, sleep / Adenosine P1 receptors / positive regulation of acetylcholine secretion, neurotransmission / G protein-coupled adenosine receptor activity / response to purine-containing compound / G protein-coupled adenosine receptor signaling pathway / NGF-independant TRKA activation / Surfactant metabolism / type 5 metabotropic glutamate receptor binding / negative regulation of vascular permeability / positive regulation of urine volume / intermediate filament / response to caffeine / blood circulation / presynaptic active zone / synaptic transmission, cholinergic / sensory perception / eating behavior / positive regulation of glutamate secretion / regulation of calcium ion transport / alpha-actinin binding / synaptic transmission, dopaminergic / membrane depolarization / asymmetric synapse / axolemma / prepulse inhibition / viral release from host cell by cytolysis / cellular defense response / phagocytosis / peptidoglycan catabolic process / neuron projection morphogenesis / positive regulation of synaptic transmission, glutamatergic / presynaptic modulation of chemical synaptic transmission / astrocyte activation / regulation of mitochondrial membrane potential / excitatory postsynaptic potential / positive regulation of protein secretion / locomotory behavior / positive regulation of long-term synaptic potentiation / central nervous system development / synaptic transmission, glutamatergic / vasodilation / cell wall macromolecule catabolic process / blood coagulation / adenylate cyclase-modulating G protein-coupled receptor signaling pathway / phospholipase C-activating G protein-coupled receptor signaling pathway / lysozyme / lysozyme activity / cell-cell signaling / adenylate cyclase-activating G protein-coupled receptor signaling pathway / presynaptic membrane / negative regulation of neuron apoptotic process / G alpha (s) signalling events / host cell cytoplasm / positive regulation of ERK1 and ERK2 cascade / calmodulin binding / postsynaptic membrane / defense response to bacterium / response to xenobiotic stimulus / inflammatory response / negative regulation of cell population proliferation / neuronal cell body / apoptotic process / regulation of DNA-templated transcription / lipid binding / dendrite / protein-containing complex binding / glutamatergic synapse / enzyme binding / membrane / identical protein binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) Enterobacteria phage T4 (virus) Enterobacteria phage T4 (virus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Jaakola, V.-P. / Griffith, M.T. / Hanson, M.A. / Cherezov, V. / Chien, E.Y.T. / Lane, J.R. / Ijzerman, A.P. / Stevens, R.C. / Accelerated Technologies Center for Gene to 3D Structure (ATCG3D) / GPCR Network (GPCR) | ||||||
 Citation Citation | Journal: Science / Year: 2008 Title: The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Authors: Jaakola, V.P. / Griffith, M.T. / Hanson, M.A. / Cherezov, V. / Chien, E.Y. / Lane, J.R. / Ijzerman, A.P. / Stevens, R.C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3eml.cif.gz | 109 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3eml.ent.gz | 81.9 KB | Display | PDB format |
| PDBx/mmJSON format | 3eml.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/em/3emlftp://data.pdbj.org/pub/pdb/validation_reports/em/3eml | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2rh1S S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Other databases |










-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 54739.770 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human), (gene. exp.) Enterobacteria phage T4 (virus)Plasmid: pBac5b / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: P29274, UniProt: P00720 Spodoptera frugiperda (fall armyworm) / References: UniProt: P29274, UniProt: P00720 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-ZMA / 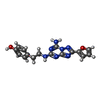  Mass: 337.336 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H15N7O2 / Comment: antagonist*YM Mass: 337.336 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H15N7O2 / Comment: antagonist*YM | ||||||
| #3: Chemical | ChemComp-STE /   Mass: 284.477 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C18H36O2 Mass: 284.477 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C18H36O2#4: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: SO4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 76 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 76 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 13 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.85 Å3/Da / Density % sol: 56.79 % Description: THIS STRUCTURE IS A PART OF THE ROADMAP/PSI COMMUNITY OUTREACH PROGRAM, NOT A SPECIFIC PSI TARGET. |
|---|---|
| Crystal grow | Temperature: 293 K / pH: 6.5 Details: PEG400 30%v/v, LiSO4 185mM, NaCitrate 100mM, pH 6.5, Lipidic mesophase, temperature 293K |
-Data collection
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 23-ID-B / Wavelength: 1.0332 / Beamline: 23-ID-B / Wavelength: 1.0332 |
|---|---|
| Detector | Type: MARMOSAIC 300 mm CCD / Detector: CCD / Date: Jun 28, 2008 / Details: MIRRORS |
| Radiation | Monochromator: DOUBLE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0332 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→20 Å / Num. obs: 18465 / % possible obs: 96.8 % / Redundancy: 3.4 % / Rsym value: 0.098 / Net I/σ(I): 7.35 |
| Reflection shell | Resolution: 2.6→2.8 Å / Redundancy: 2.3 % / Mean I/σ(I) obs: 2.3 / Rsym value: 0.398 / % possible all: 93.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2RH1 Resolution: 2.6→19.42 Å / SU ML: 0.43 / Isotropic thermal model: Isotropic / σ(F): 2 / Stereochemistry target values: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 78.49 Å2 / ksol: 0.33 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→19.42 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
