phosphorelay sensor kinase activity / histidine kinase / phosphoprotein phosphatase activity / protein kinase activity / protein dimerization activity / ATP binding / identical protein binding / plasma membrane Similarity search - Function
Single alpha-helices involved in coiled-coils or other helix-helix interfaces - #2870 / Single alpha-helices involved in coiled-coils or other helix-helix interfaces - #1930 / : / DesK N-terminal domain / Signal transduction histidine kinase, subgroup 3, dimerisation and phosphoacceptor domain / : / Histidine kinase / Single alpha-helices involved in coiled-coils or other helix-helix interfaces / Histidine kinase-like ATPase, C-terminal domain / Heat Shock Protein 90 ...Single alpha-helices involved in coiled-coils or other helix-helix interfaces - #2870 / Single alpha-helices involved in coiled-coils or other helix-helix interfaces - #1930 / : / DesK N-terminal domain / Signal transduction histidine kinase, subgroup 3, dimerisation and phosphoacceptor domain / : / Histidine kinase / Single alpha-helices involved in coiled-coils or other helix-helix interfaces / Histidine kinase-like ATPase, C-terminal domain / Heat Shock Protein 90 / Histidine kinase-, DNA gyrase B-, and HSP90-like ATPase / Single alpha-helices involved in coiled-coils or other helix-helix interfaces / Helix non-globular / Special / Histidine kinase/HSP90-like ATPase superfamily / Up-down Bundle / 2-Layer Sandwich / Mainly Alpha / Alpha Beta Similarity search - Domain/homology
Monochromator: channel-cut / Protocol: SAD / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.9793 Å / Relative weight: 1
Reflection
Resolution: 3.1→26.7 Å / Num. obs: 15927 / % possible obs: 99.1 % / Observed criterion σ(I): 0 / Biso Wilson estimate: 54.042 Å2 / Rmerge(I) obs: 0.11
Reflection shell
Resolution (Å)
Highest resolution (Å)
Rmerge(I) obs
Mean I/σ(I) obs
Num. measured obs
Num. unique obs
% possible all
3.1-3.2
0.444
7.1
19418
1439
99.4
3.2-3.3
0.399
8.3
16918
1246
99
3.3-3.4
0.298
11.5
15574
1125
99.6
3.4-3.5
0.242
14.4
13895
982
99.1
3.5-4
0.175
19.5
52779
3651
99.5
4-6
0.091
29.7
75236
5196
99.2
6-10
0.059
37.2
24667
1770
98.7
10-20
0.046
44.2
5825
451
98.3
20
0.058
25.8
339
67
90.5
-
Phasing
Phasing
Method: SAD
Phasing MAD set
ID
R cullis acentric
R cullis centric
Highest resolution (Å)
Lowest resolution (Å)
Reflection acentric
Reflection centric
ISO_1
0
0
3.5
43.73
9775
1116
ANO_1
0.814
0
3.5
43.73
9769
0
Phasing MAD set shell
ID
Resolution (Å)
R cullis acentric
R cullis centric
Reflection acentric
Reflection centric
ISO_1
14.78-43.73
0
0
98
58
ISO_1
10.76-14.78
0
0
188
59
ISO_1
8.88-10.76
0
0
232
53
ISO_1
7.73-8.88
0
0
302
54
ISO_1
6.93-7.73
0
0
337
56
ISO_1
6.34-6.93
0
0
373
58
ISO_1
5.88-6.34
0
0
414
60
ISO_1
5.51-5.88
0
0
442
53
ISO_1
5.2-5.51
0
0
499
59
ISO_1
4.93-5.2
0
0
472
59
ISO_1
4.71-4.93
0
0
553
50
ISO_1
4.51-4.71
0
0
539
57
ISO_1
4.33-4.51
0
0
591
56
ISO_1
4.18-4.33
0
0
590
49
ISO_1
4.04-4.18
0
0
626
46
ISO_1
3.91-4.04
0
0
683
63
ISO_1
3.79-3.91
0
0
664
49
ISO_1
3.69-3.79
0
0
697
58
ISO_1
3.59-3.69
0
0
740
62
ISO_1
3.5-3.59
0
0
735
57
ANO_1
14.78-43.73
0.392
0
98
0
ANO_1
10.76-14.78
0.595
0
188
0
ANO_1
8.88-10.76
0.549
0
232
0
ANO_1
7.73-8.88
0.62
0
301
0
ANO_1
6.93-7.73
0.618
0
337
0
ANO_1
6.34-6.93
0.669
0
373
0
ANO_1
5.88-6.34
0.737
0
413
0
ANO_1
5.51-5.88
0.725
0
442
0
ANO_1
5.2-5.51
0.814
0
499
0
ANO_1
4.93-5.2
0.816
0
471
0
ANO_1
4.71-4.93
0.827
0
551
0
ANO_1
4.51-4.71
0.875
0
539
0
ANO_1
4.33-4.51
0.853
0
591
0
ANO_1
4.18-4.33
0.899
0
590
0
ANO_1
4.04-4.18
0.961
0
625
0
ANO_1
3.91-4.04
0.955
0
683
0
ANO_1
3.79-3.91
0.974
0
664
0
ANO_1
3.69-3.79
0.997
0
697
0
ANO_1
3.59-3.69
0.976
0
740
0
ANO_1
3.5-3.59
0.984
0
735
0
Phasing MAD set site
ID
Cartn x (Å)
Cartn y (Å)
Cartn z (Å)
Atom type symbol
B iso
Occupancy
1
51.262
26.368
15.051
SE
154.98
2.39
2
17.852
24.905
-5.685
SE
138.85
1.46
3
39.187
21.823
12.289
SE
42.23
0.46
4
36.519
24.336
15.306
SE
147.87
0.25
5
29.989
17.487
11.964
SE
155.32
2.05
6
28.352
22.389
12.548
SE
131.66
1.83
7
53.092
23.853
19.406
SE
137.45
1.64
8
58.462
30.44
27.688
SE
53.85
0.43
9
59.896
38.656
31.208
SE
67.91
0.76
10
34.472
35.937
78.383
SE
159.42
1.48
11
38.561
33.433
79.844
SE
145.48
1.47
12
49.907
49.421
111.014
SE
103.73
2.05
13
37.43
41.296
90.183
SE
39.08
0.63
14
32.453
47.931
97.071
SE
114.79
1.67
15
33.392
43.34
100.151
SE
126.15
1.87
Phasing dm
Method: Solvent flattening and Histogram matching / Reflection: 23982
Phasing dm shell
Resolution (Å)
Delta phi final
FOM
Reflection
10.13-100
47.7
0.74
505
7.96-10.13
40.7
0.87
503
6.9-7.96
42.4
0.884
512
6.24-6.9
39.4
0.878
513
5.74-6.24
38.1
0.894
579
5.34-5.74
34.2
0.903
606
5.02-5.34
34.4
0.895
640
4.75-5.02
27
0.907
686
4.52-4.75
27.3
0.908
693
4.32-4.52
25.5
0.911
776
4.14-4.32
28.6
0.914
768
3.98-4.14
26.3
0.92
804
3.84-3.98
24.6
0.932
863
3.72-3.84
24.3
0.931
874
3.6-3.72
24.9
0.92
901
3.5-3.6
23.3
0.92
923
3.4-3.5
25.9
0.912
932
3.31-3.4
25.4
0.914
1000
3.23-3.31
27.5
0.903
998
3.16-3.23
29
0.903
1010
3.09-3.16
28.2
0.903
1078
3.02-3.09
30.3
0.897
1058
2.96-3.02
29.6
0.891
1085
2.9-2.96
31
0.882
1130
2.84-2.9
34.5
0.87
1124
2.79-2.84
37.8
0.852
1162
2.7-2.79
44
0.807
2259
-
Processing
Software
Name
Version
Classification
NB
XSCALE
datascaling
SHARP
phasing
DM
6
phasing
REFMAC
5.5.0053
refinement
PDB_EXTRACT
3.006
dataextraction
Refinement
Method to determine structure: SAD / Resolution: 3.1→15 Å / Cor.coef. Fo:Fc: 0.901 / Cor.coef. Fo:Fc free: 0.874 / Occupancy max: 1 / Occupancy min: 0.29 / SU B: 43.355 / SU ML: 0.379 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.488 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: Hydrogens have been added in the riding positions atom record contains sum of TLS and residual B anisou record contains sum of TLS and residual U
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.297
816
5.2 %
RANDOM
Rwork
0.242
14957
-
-
obs
0.245
15773
99.24 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads













 PDBj
PDBj




 Assembly
Assembly


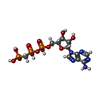
 Mass: 505.208 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM
Mass: 505.208 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM
 Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID14-4 / Wavelength: 0.9793 Å
/ Beamline: ID14-4 / Wavelength: 0.9793 Å Processing
Processing