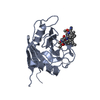
- PDB-3cfy: Crystal structure of signal receiver domain of putative Luxo repr... -
+
データを開く
IDまたはキーワード:
読み込み中...
-
基本情報
登録情報
データベース: PDB / ID: 3cfy
タイトル
Crystal structure of signal receiver domain of putative Luxo repressor protein from Vibrio parahaemolyticus
要素
Putative LuxO repressor protein
キーワード
STRUCTURAL GENOMICS / UNKNOWN FUNCTION / UNCHARACTERIZED PROTEIN / SIGNAL RECEIVER DOMAIN / AAA-ATPASE / PROTEIN STRUCTURE INITIATIVE / PSI-2 / New York SGX Research Center for Structural Genomics / NYSGXRC / ATP-binding / DNA-binding / Nucleotide-binding / Transcription / Transcription regulation
機能・相同性
機能・相同性情報
phosphorelay signal transduction system / sequence-specific DNA binding / regulation of DNA-templated transcription / ATP hydrolysis activity / ATP binding 類似検索 - 分子機能
Sigma-54 interaction domain, conserved site / Sigma-54 interaction domain C-terminal part signature. / Sigma-54 interaction domain, ATP-binding site 2 / Sigma-54 interaction domain ATP-binding region B signature. / Sigma-54 interaction domain profile. / Sigma-54 interaction domain / RNA polymerase sigma factor 54 interaction domain / DNA binding HTH domain, Fis-type / Bacterial regulatory protein, Fis family / Response regulator receiver domain ...Sigma-54 interaction domain, conserved site / Sigma-54 interaction domain C-terminal part signature. / Sigma-54 interaction domain, ATP-binding site 2 / Sigma-54 interaction domain ATP-binding region B signature. / Sigma-54 interaction domain profile. / Sigma-54 interaction domain / RNA polymerase sigma factor 54 interaction domain / DNA binding HTH domain, Fis-type / Bacterial regulatory protein, Fis family / Response regulator receiver domain / cheY-homologous receiver domain / Signal transduction response regulator, receiver domain / Response regulatory domain profile. / CheY-like superfamily / Response regulator / Homeobox-like domain superfamily / ATPases associated with a variety of cellular activities / AAA+ ATPase domain / Rossmann fold / P-loop containing nucleoside triphosphate hydrolase / 3-Layer(aba) Sandwich / Alpha Beta 類似検索 - ドメイン・相同性
モノクロメーター: DIAMOND / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray
放射波長
波長: 0.9793 Å / 相対比: 1
反射
解像度: 2.25→50 Å / Num. obs: 8444 / % possible obs: 99.3 % / Observed criterion σ(I): -0.5 / 冗長度: 7.4 % / Rmerge(I) obs: 0.061 / Net I/σ(I): 6.1
反射 シェル
解像度: 2.25→2.33 Å / 冗長度: 5.6 % / Rmerge(I) obs: 0.82 / Mean I/σ(I) obs: 1.1 / % possible all: 98.4
-
解析
ソフトウェア
名称
バージョン
分類
SHELX
モデル構築
REFMAC
5.3.0034
精密化
MAR345
CCD
データ収集
HKL-2000
データ削減
HKL-2000
データスケーリング
SHELXD
位相決定
精密化
構造決定の手法: 単波長異常分散 / 解像度: 2.5→20 Å / Cor.coef. Fo:Fc: 0.941 / Cor.coef. Fo:Fc free: 0.912 / SU B: 15.227 / SU ML: 0.297 / 交差検証法: THROUGHOUT / ESU R: 0.489 / ESU R Free: 0.31 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD / 詳細: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
反射数
%反射
Selection details
Rfree
0.2907
167
2.7 %
RANDOM
Rwork
0.24645
-
-
-
obs
0.24763
5965
99.46 %
-
溶媒の処理
イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.2 Å / 溶媒モデル: MASK
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Vibrio parahaemolyticus RIMD 2210633 (腸炎ビブリオ)
Vibrio parahaemolyticus RIMD 2210633 (腸炎ビブリオ) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード







 PDBj
PDBj

 集合体
集合体
 分子量: 18.015 Da / 分子数: 18 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 18 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 31-ID / 波長: 0.9793 Å
/ ビームライン: 31-ID / 波長: 0.9793 Å 解析
解析