Mass: 18.015 Da / Num. of mol.: 96 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
ID
Density Matthews (Å3/Da)
Density % sol (%)
1
2.63
53.25
2
Crystal grow
Crystal-ID
Details
1
Native crystal, used for model refinement: 26% PEG 4000, 0.1M Tris-HCl, 0.2M Magnesium chloride, 0.1mM DTT, pH 8.0, VAPOR DIFFUSION, SITTING DROP, temperature 291K
2
Seleno-methionine derivative crystal, used for MAD phasing: 33% PEG 4000, 0.1M Tris-HCl, 0.2M Magnesium chloride, 0.1mM DTT, pH 9.0, VAPOR DIFFUSION, SITTING DROP, temperature 291K. A ten-fold excess of GppNHp was added to the protein samples prior to crystallization.
-
Data collection
Diffraction
ID
Mean temperature (K)
Crystal-ID
1
100
1
2
100
1
Diffraction source
Source
Site
Beamline
ID
Wavelength (Å)
SYNCHROTRON
APS
23-ID-B
1
0.97926
SYNCHROTRON
APS
19-ID
2
0.97931, 0.97945, 0.98166, 0.97243
Detector
Type
ID
Detector
Date
MARMOSAIC 300 mm CCD
1
CCD
Nov 29, 2007
ADSC QUANTUM 315
2
CCD
Dec 6, 2007
Radiation
ID
Protocol
Monochromatic (M) / Laue (L)
Scattering type
Wavelength-ID
1
SINGLEWAVELENGTH
M
x-ray
1
2
MAD
M
x-ray
1
Radiation wavelength
ID
Wavelength (Å)
Relative weight
1
0.97926
1
2
0.97931
1
3
0.97945
1
4
0.98166
1
5
0.97243
1
Reflection
Resolution: 1.8→50 Å / Num. obs: 26661 / % possible obs: 95.1 % / Redundancy: 6.7 % / Rmerge(I) obs: 0.075 / Χ2: 1.527 / Net I/σ(I): 10.9
Reflection shell
Resolution (Å)
Redundancy (%)
Rmerge(I) obs
Num. unique all
Χ2
% possible all
1.8-1.86
2.7
0.456
1690
0.941
61.8
1.86-1.94
4.1
0.387
2491
1.14
89.1
1.94-2.03
6.1
0.328
2796
1.053
98.9
2.03-2.13
7.3
0.238
2788
1.184
100
2.13-2.27
7.5
0.18
2797
1.296
100
2.27-2.44
7.6
0.14
2791
1.306
100
2.44-2.69
7.6
0.103
2775
1.331
100
2.69-3.08
7.6
0.08
2811
1.779
100
3.08-3.88
7.5
0.055
2839
2.219
100
3.88-50
7.4
0.045
2883
2.174
100
-
Phasing
Phasing
Method: mad
Phasing set
ID
1
2
3
4
Phasing MAD
D res high: 3 Å / D res low: 30 Å / FOM : 0.71 / Reflection: 6679
Method to determine structure: MAD / Resolution: 1.8→19.26 Å / Cor.coef. Fo:Fc: 0.95 / Cor.coef. Fo:Fc free: 0.926 / WRfactor Rfree: 0.238 / WRfactor Rwork: 0.197 / SU B: 2.936 / SU ML: 0.092 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.127 / ESU R Free: 0.127 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: Due to considerable non-isomorphism between the phasing and refinement data sets, a preliminary model obtained by the program 'resolve' based on the phasing data was used in molecular ...Details: Due to considerable non-isomorphism between the phasing and refinement data sets, a preliminary model obtained by the program 'resolve' based on the phasing data was used in molecular replacement with the higher resolution refinement data and the program 'phaser'. Following density modification with 'DM', 'ARP/wARP' was used for autotracing of the higher resolution model. HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS during final refinement. Programs, ARP/wARP, coot, molprobity have also been used in refinement
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.247
1352
5.1 %
RANDOM
Rwork
0.205
-
-
-
obs
0.207
26626
95.06 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK
Displacement parameters
Biso mean: 21.063 Å2
Baniso -1
Baniso -2
Baniso -3
1-
0.02 Å2
0 Å2
0 Å2
2-
-
0.02 Å2
0 Å2
3-
-
-
-0.03 Å2
Refinement step
Cycle: LAST / Resolution: 1.8→19.26 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
1799
0
39
96
1934
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.017
0.022
1869
X-RAY DIFFRACTION
r_bond_other_d
0.001
0.02
1193
X-RAY DIFFRACTION
r_angle_refined_deg
1.465
1.973
2549
X-RAY DIFFRACTION
r_angle_other_deg
0.954
3
2938
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
6.285
5
238
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
33.331
25.309
81
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
11.656
15
306
X-RAY DIFFRACTION
r_dihedral_angle_4_deg
25.159
15
5
X-RAY DIFFRACTION
r_chiral_restr
0.086
0.2
298
X-RAY DIFFRACTION
r_gen_planes_refined
0.005
0.02
2076
X-RAY DIFFRACTION
r_gen_planes_other
0.001
0.02
364
X-RAY DIFFRACTION
r_nbd_refined
0.211
0.2
352
X-RAY DIFFRACTION
r_nbd_other
0.186
0.2
1235
X-RAY DIFFRACTION
r_nbtor_refined
0.173
0.2
916
X-RAY DIFFRACTION
r_nbtor_other
0.081
0.2
915
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.131
0.2
94
X-RAY DIFFRACTION
r_metal_ion_refined
0.027
0.2
2
X-RAY DIFFRACTION
r_symmetry_vdw_refined
0.284
0.2
9
X-RAY DIFFRACTION
r_symmetry_vdw_other
0.229
0.2
18
X-RAY DIFFRACTION
r_symmetry_hbond_refined
0.119
0.2
7
X-RAY DIFFRACTION
r_mcbond_it
2.72
2
1291
X-RAY DIFFRACTION
r_mcbond_other
0.702
2
477
X-RAY DIFFRACTION
r_mcangle_it
3.601
3
1876
X-RAY DIFFRACTION
r_scbond_it
2.861
2
785
X-RAY DIFFRACTION
r_scangle_it
3.899
3
668
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Num. reflection all
% reflection obs (%)
1.8-1.848
0.391
61
0.285
1152
2089
58.066
1.848-1.898
0.298
75
0.267
1501
1952
80.738
1.898-1.952
0.344
95
0.249
1718
1923
94.28
1.952-2.012
0.277
79
0.231
1779
1875
99.093
2.012-2.077
0.302
94
0.226
1772
1867
99.946
2.077-2.149
0.237
108
0.208
1653
1761
100
2.149-2.229
0.292
83
0.21
1615
1698
100
2.229-2.319
0.24
90
0.203
1567
1659
99.879
2.319-2.42
0.257
84
0.211
1482
1566
100
2.42-2.536
0.222
76
0.205
1452
1528
100
2.536-2.671
0.237
79
0.207
1348
1427
100
2.671-2.829
0.217
77
0.209
1273
1350
100
2.829-3.02
0.339
55
0.208
1240
1296
99.923
3.02-3.255
0.239
66
0.202
1145
1211
100
3.255-3.556
0.242
45
0.196
1058
1104
99.909
3.556-3.959
0.246
53
0.186
948
1001
100
3.959-4.54
0.193
43
0.16
870
913
100
4.54-5.484
0.196
38
0.177
726
764
100
5.484-7.459
0.229
26
0.244
602
628
100
7.459-19.263
0.245
25
0.214
373
399
99.749
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj




 Assembly
Assembly


 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg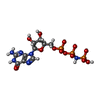
 Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3
Mass: 522.196 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N6O13P3
 Num. of mol.: 5 / Source method: obtained synthetically
Num. of mol.: 5 / Source method: obtained synthetically Mass: 18.015 Da / Num. of mol.: 96 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 96 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation
 Processing
Processing