phosphorelay sensor kinase activity / histidine kinase / signal transduction / ATP binding / metal ion binding / plasma membrane Similarity search - Function
Signal transduction histidine kinase, DctB (C4-dicarboxylate transport system regulator) / Cache domain / Periplasmic sensor-like domain superfamily / His Kinase A (phospho-acceptor) domain / His Kinase A (phosphoacceptor) domain / Signal transduction histidine kinase, dimerisation/phosphoacceptor domain / PAS domain / Signal transduction histidine kinase-related protein, C-terminal / Signal transduction histidine kinase, dimerisation/phosphoacceptor domain superfamily / Histidine kinase domain ...Signal transduction histidine kinase, DctB (C4-dicarboxylate transport system regulator) / Cache domain / Periplasmic sensor-like domain superfamily / His Kinase A (phospho-acceptor) domain / His Kinase A (phosphoacceptor) domain / Signal transduction histidine kinase, dimerisation/phosphoacceptor domain / PAS domain / Signal transduction histidine kinase-related protein, C-terminal / Signal transduction histidine kinase, dimerisation/phosphoacceptor domain superfamily / Histidine kinase domain / Histidine kinase domain profile. / Beta-Lactamase / Histidine kinase-, DNA gyrase B-, and HSP90-like ATPase / Histidine kinase-like ATPases / Histidine kinase/HSP90-like ATPase / Histidine kinase/HSP90-like ATPase superfamily / 2-Layer Sandwich / Alpha Beta Similarity search - Domain/homology
Av σ(I) over netI: 15.4 / Number: 284212 / Rmerge(I) obs: 0.061 / Χ2: 1.01 / D res high: 2.2 Å / D res low: 20 Å / Num. obs: 56978 / % possible obs: 98.8
Diffraction reflection shell
Highest resolution (Å)
Lowest resolution (Å)
% possible obs (%)
ID
Rmerge(I) obs
Chi squared
4.72
20
99.9
1
0.041
1.01
3.76
4.72
100
1
0.044
1.026
3.28
3.76
100
1
0.051
1.02
2.98
3.28
100
1
0.062
1.023
2.77
2.98
100
1
0.075
1.04
2.61
2.77
100
1
0.092
1.033
2.48
2.61
99.9
1
0.106
1.027
2.37
2.48
99.4
1
0.116
0.994
2.28
2.37
97.7
1
0.125
0.947
2.2
2.28
91.2
1
0.129
0.914
Reflection
Resolution: 1.6→20 Å / Num. obs: 76821 / % possible obs: 98.4 % / Rmerge(I) obs: 0.057 / Χ2: 0.998 / Net I/σ(I): 11.5
Reflection shell
Resolution (Å)
Rmerge(I) obs
Num. unique all
Χ2
% possible all
1.6-1.66
0.395
7089
0.83
91.8
1.66-1.72
0.314
7419
0.885
96.5
1.72-1.8
0.237
7698
0.966
99.7
1.8-1.9
0.176
7707
1.022
99.6
1.9-2.02
0.123
7695
1.041
99.5
2.02-2.17
0.089
7738
1.032
99.6
2.17-2.39
0.07
7748
1.018
99.7
2.39-2.73
0.056
7793
1.03
99.6
2.73-3.44
0.039
7853
1.043
99.4
3.44-20
0.032
8081
1.03
98.6
-
Phasing
Phasing
Method: MAD
Phasing dm
Method: Solvent flattening and Histogram matching / Reflection: 64840
Phasing dm shell
Resolution (Å)
Delta phi final
FOM
Reflection
8.6-100
46.3
0.927
506
6.29-8.6
42.3
0.936
834
5.19-6.29
39.2
0.938
1041
4.53-5.19
37.9
0.949
1221
4.06-4.53
39.2
0.943
1342
3.72-4.06
41.4
0.936
1476
3.45-3.72
42.1
0.924
1609
3.23-3.45
42.6
0.918
1709
3.05-3.23
42.7
0.916
1808
2.89-3.05
44.3
0.899
1917
2.76-2.89
44.5
0.909
2001
2.64-2.76
44.2
0.907
2101
2.54-2.64
46.8
0.904
2176
2.45-2.54
51.7
0.901
2241
2.37-2.45
54.3
0.908
2361
2.29-2.37
60.2
0.914
2418
2.23-2.29
64.6
0.905
2493
2.16-2.23
81.2
0.902
2544
2.11-2.16
90.7
0.908
2638
2.05-2.11
90.2
0.904
2691
2-2.05
88.3
0.902
2741
1.96-2
89.5
0.899
2820
1.92-1.96
89
0.886
2926
1.88-1.92
91
0.885
2897
1.84-1.88
89
0.876
3021
1.8-1.84
90
0.874
3059
1.77-1.8
90.9
0.859
3123
1.74-1.77
90.4
0.815
3190
1.7-1.74
90
0.677
3936
-
Processing
Software
Name
Version
Classification
NB
DENZO
datareduction
SCALEPACK
datascaling
SOLVE
phasing
DM
4.2
phasing
CNS
1.1
refinement
PDB_EXTRACT
3.004
dataextraction
Refinement
Method to determine structure: MAD / Resolution: 1.7→19.78 Å / Rfactor Rfree error: 0.003 / FOM work R set: 0.895 / Data cutoff high absF: 1911155.375 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Vibrio cholerae (bacteria)
Vibrio cholerae (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj

 Assembly
Assembly



 Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca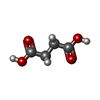
 Mass: 118.088 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O4
Mass: 118.088 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O4 Mass: 18.015 Da / Num. of mol.: 721 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 721 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: X4A / Wavelength: 0.9637, 0.9788, 0.9791, 0.9946
/ Beamline: X4A / Wavelength: 0.9637, 0.9788, 0.9791, 0.9946 Processing
Processing