| ソフトウェア | | 名称 | バージョン | 分類 | NB |
|---|
| DENZO | | データ削減 | | | SCALEPACK | | データスケーリング | | | PHASER | | 位相決定 | | | REFMAC | | 精密化 | | | PDB_EXTRACT | 3.004 | データ抽出 | | | MX-based | | データ収集 | |
|
|---|
| 精密化 | 構造決定の手法:  分子置換 / 解像度: 2→79.31 Å / Cor.coef. Fo:Fc: 0.95 / Cor.coef. Fo:Fc free: 0.931 / SU B: 11.061 / SU ML: 0.155 / TLS residual ADP flag: LIKELY RESIDUAL / 交差検証法: THROUGHOUT / σ(F): 0 / ESU R: 0.194 / ESU R Free: 0.173 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD / 詳細: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS 分子置換 / 解像度: 2→79.31 Å / Cor.coef. Fo:Fc: 0.95 / Cor.coef. Fo:Fc free: 0.931 / SU B: 11.061 / SU ML: 0.155 / TLS residual ADP flag: LIKELY RESIDUAL / 交差検証法: THROUGHOUT / σ(F): 0 / ESU R: 0.194 / ESU R Free: 0.173 / 立体化学のターゲット値: MAXIMUM LIKELIHOOD / 詳細: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| Rfactor | 反射数 | %反射 | Selection details |
|---|
| Rfree | 0.26 | 1206 | 5 % | RANDOM |
|---|
| Rwork | 0.222 | - | - | - |
|---|
| obs | 0.224 | 23961 | 99.69 % | - |
|---|
|
|---|
| 溶媒の処理 | イオンプローブ半径: 0.8 Å / 減衰半径: 0.8 Å / VDWプローブ半径: 1.2 Å / 溶媒モデル: MASK |
|---|
| 原子変位パラメータ | Biso mean: 43.938 Å2
| Baniso -1 | Baniso -2 | Baniso -3 |
|---|
| 1- | -1.34 Å2 | -0.67 Å2 | 0 Å2 |
|---|
| 2- | - | -1.34 Å2 | 0 Å2 |
|---|
| 3- | - | - | 2 Å2 |
|---|
|
|---|
| 精密化ステップ | サイクル: LAST / 解像度: 2→79.31 Å
| タンパク質 | 核酸 | リガンド | 溶媒 | 全体 |
|---|
| 原子数 | 2285 | 0 | 26 | 79 | 2390 |
|---|
|
|---|
| 拘束条件 | | Refine-ID | タイプ | Dev ideal | Dev ideal target | 数 |
|---|
| X-RAY DIFFRACTION | r_bond_refined_d| 0.013 | 0.022 | 2385 | | X-RAY DIFFRACTION | r_angle_refined_deg| 1.214 | 1.96 | 3254 | | X-RAY DIFFRACTION | r_dihedral_angle_1_deg| 5.595 | 5 | 299 | | X-RAY DIFFRACTION | r_dihedral_angle_2_deg| 36.475 | 24.706 | 102 | | X-RAY DIFFRACTION | r_dihedral_angle_3_deg| 14.027 | 15 | 347 | | X-RAY DIFFRACTION | r_dihedral_angle_4_deg| 13.722 | 15 | 8 | | X-RAY DIFFRACTION | r_chiral_restr| 0.373 | 0.2 | 345 | | X-RAY DIFFRACTION | r_gen_planes_refined| 0.016 | 0.02 | 1860 | | X-RAY DIFFRACTION | r_nbd_refined| 0.255 | 0.2 | 994 | | X-RAY DIFFRACTION | r_nbtor_refined| 0.323 | 0.2 | 1629 | | X-RAY DIFFRACTION | r_xyhbond_nbd_refined| 0.21 | 0.2 | 106 | | X-RAY DIFFRACTION | r_symmetry_vdw_refined| 0.326 | 0.2 | 43 | | X-RAY DIFFRACTION | r_symmetry_hbond_refined| 0.227 | 0.2 | 7 | | X-RAY DIFFRACTION | r_mcbond_it| 3.378 | 1.5 | 1519 | | X-RAY DIFFRACTION | r_mcangle_it| 4.691 | 2 | 2407 | | X-RAY DIFFRACTION | r_scbond_it| 6.911 | 3 | 1009 | | X-RAY DIFFRACTION | r_scangle_it| 8.749 | 4.5 | 847 | | | | | | | | | | | | | | | | | |
|
|---|
| LS精密化 シェル | 解像度: 2→2.052 Å / Total num. of bins used: 20
| Rfactor | 反射数 | %反射 |
|---|
| Rfree | 0.414 | 81 | - |
|---|
| Rwork | 0.286 | 1680 | - |
|---|
| all | - | 1761 | - |
|---|
| obs | - | - | 100 % |
|---|
|
|---|
| 精密化 TLS | 手法: refined / Origin x: 0.0904 Å / Origin y: 26.5729 Å / Origin z: -4.6386 Å
| 11 | 12 | 13 | 21 | 22 | 23 | 31 | 32 | 33 |
|---|
| T | -0.003 Å2 | -0.018 Å2 | 0.1649 Å2 | - | -0.194 Å2 | -0.0114 Å2 | - | - | -0.0329 Å2 |
|---|
| L | 0.7861 °2 | -1.0606 °2 | 1.0214 °2 | - | 2.6487 °2 | -0.2327 °2 | - | - | 2.454 °2 |
|---|
| S | -0.0513 Å ° | 0.0671 Å ° | -0.1012 Å ° | -0.0319 Å ° | 0.0651 Å ° | -0.1038 Å ° | 0.5548 Å ° | 0.0272 Å ° | -0.0138 Å ° |
|---|
|
|---|
| 精密化 TLSグループ | | ID | Refine-ID | Refine TLS-ID | Auth asym-ID | Label asym-ID | Auth seq-ID | Label seq-ID |
|---|
| 1 | X-RAY DIFFRACTION | 1 | AA| 6 - 157 | 6 - 157 | | 2 | X-RAY DIFFRACTION | 1 | BB| 6 - 157 | 6 - 157 | | | | |
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Argas monolakensis (ダニ)
Argas monolakensis (ダニ) データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード












 PDBj
PDBj

 集合体
集合体



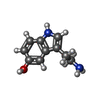
 分子量: 176.215 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C10H12N2O / コメント: 神経伝達物質*YM
分子量: 176.215 Da / 分子数: 2 / 由来タイプ: 合成 / 式: C10H12N2O / コメント: 神経伝達物質*YM 分子量: 18.015 Da / 分子数: 79 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 79 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: 22-BM / 波長: 1 Å
/ ビームライン: 22-BM / 波長: 1 Å 解析
解析