 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3bnr: Crystal Structure of the Homo sapiens Mitochondrial Ribosomal Dec... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3bnr | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Homo sapiens Mitochondrial Ribosomal Decoding Site in the presence of nonspecifically bound paromomycin (A1555G mutant, Br-derivative) | ||||||
 Components Components | (A site of human mitochondrial ribosome, chain ...) x 3 | ||||||
 Keywords Keywords | RNA / ribosome / decoding site | ||||||
| Function / homology | : / PAROMOMYCIN / RNA / RNA (> 10) Function and homology information Function and homology information | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.1 Å | ||||||
 Authors Authors | Kondo, J. / Westhof, E. | ||||||
 Citation Citation | Journal: Nucleic Acids Res. / Year: 2008 Title: The bacterial and mitochondrial ribosomal A-site molecular switches possess different conformational substates Authors: Kondo, J. / Westhof, E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3bnr.cif.gz | 65.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3bnr.ent.gz | 47.6 KB | Display | PDB format |
| PDBx/mmJSON format | 3bnr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bn/3bnrftp://data.pdbj.org/pub/pdb/validation_reports/bn/3bnr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3bnlC  3bnnC  3bnoC  3bnpC  3bnqC  3bnsC  3bntC C: citing same article ( |
|---|---|
| Similar structure data |

-Links
 PDBj
PDBj





























- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-A site of human mitochondrial ribosome, chain ... , 3 types, 4 molecules ACBD
| #1: RNA chain | Mass: 7394.280 Da / Num. of mol.: 2 / Mutation: A1555G / Source method: obtained synthetically #2: RNA chain | | Mass: 7088.115 Da / Num. of mol.: 1 / Mutation: A1555G / Source method: obtained synthetically #3: RNA chain | | Mass: 6781.949 Da / Num. of mol.: 1 / Mutation: A1555G / Source method: obtained synthetically |
|---|
-Non-polymers , 3 types, 186 molecules 
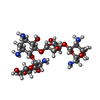

| #4: Chemical |  Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K#5: Chemical | ChemComp-PAR / |  Mass: 615.628 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H45N5O14 / Comment: Antimicrobial, medication*YM Mass: 615.628 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C23H45N5O14 / Comment: Antimicrobial, medication*YM#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 183 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.45 Å3/Da / Density % sol: 49.81 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7 Details: Sodium cacodylate, KCl, spermine tetrachloride, paromomycin, 2-methyl-2,4-pentanediol, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||
| Components of the solutions |
|
-Data collection
| Diffraction | Mean temperature: 100 K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID23-1 / Wavelength: 0.91625 Å / Beamline: ID23-1 / Wavelength: 0.91625 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MAR CCD 165 mm / Detector: CCD / Date: May 20, 2007 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.91625 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.1→50.995 Å / Num. obs: 16388 / % possible obs: 99.8 % / Redundancy: 6.9 % / Rmerge(I) obs: 0.116 / Rsym value: 0.116 / Net I/σ(I): 2.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.1→50.995 Å / FOM work R set: 0.828 / σ(F): 0
| ||||||||||||||||||||
| Solvent computation | Bsol: 69.092 Å2 | ||||||||||||||||||||
| Displacement parameters | Biso mean: 36.922 Å2
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→50.995 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Xplor file |
|
