 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-363d: High-resolution crystal structure of a fully modified N3'-> P5' p... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 363d | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | High-resolution crystal structure of a fully modified N3'-> P5' phosphoramidate DNA dodecamer duplex | ||||||||||||||||||
 Components Components | 5'-D(* Keywords KeywordsDNA / A-DNA / MODIFIED | Function / homology | AMMONIUM ION / DNA / DNA (> 10) |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å  Authors AuthorsTereshko, V. / Gryaznov, S. / Egli, M. |  Citation CitationJournal: J.Am.Chem.Soc. / Year: 1998 | Title: Consequences of Replacing the DNA 3'-Oxygen by an Amino Group: High-Resolution Crystal Structure of a Fully Modified N3'--> P5' Phosphoramidate DNA Dodecamer Duplex Authors: Tereshko, V. / Gryaznov, S. / Egli, M. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 363d.cif.gz | 57.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb363d.ent.gz | 50.5 KB | Display | PDB format |
| PDBx/mmJSON format | 363d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/63/363dftp://data.pdbj.org/pub/pdb/validation_reports/63/363d | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|





-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: DNA chain | Mass: 3652.559 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source #2: Chemical | ChemComp-NH4 / 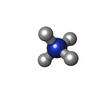  Mass: 18.038 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: H4N Mass: 18.038 Da / Num. of mol.: 24 / Source method: obtained synthetically / Formula: H4N#3: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 12 / Source method: obtained synthetically / Formula: Cl#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 200 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 200 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7 Details: pH 7.00, VAPOR DIFFUSION, HANGING DROP, temperature 293.00K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Components of the solutions |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop / PH range low: 7.4 / PH range high: 6.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 290 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Apr 1, 1996 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Relative weight: 1 |
| Reflection | Resolution: 2→34 Å / Num. obs: 6737 / % possible obs: 98.4 % / Rmerge(I) obs: 0.082 |
| Reflection shell | Highest resolution: 2 Å / Redundancy: 11.4 % |
| Reflection | *PLUS Highest resolution: 2 Å / Lowest resolution: 34 Å / % possible obs: 98.4 % / Num. measured all: 76542 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: A-FORM DUPLEX Resolution: 2→8 Å / Cross valid method: FREE R-VALUE / σ(F): 2 Details: THE CRYSTAL IS A PRECISE MEROHEDRAL TWIN AND THE COORDINATES REPRESENT ONE COLUMN CONSISTING OF THREE DODECAMER DUPLEXES (ALL ATOMS WITH OCCUPANCY 0.5, HALF THE WATERS HAVE OCCUPANCY 1.0, ...Details: THE CRYSTAL IS A PRECISE MEROHEDRAL TWIN AND THE COORDINATES REPRESENT ONE COLUMN CONSISTING OF THREE DODECAMER DUPLEXES (ALL ATOMS WITH OCCUPANCY 0.5, HALF THE WATERS HAVE OCCUPANCY 1.0, THE OTHER HALF ZERO). TOTAL NUMBER OF SOLVENT ATOMS REFERS TO R3 ASYMMETRIC UNIT.
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→8 Å
| |||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 8 Å / σ(F): 2 / % reflection Rfree: 15 % | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
