| 登録情報 | データベース: PDB / ID: 312d
|
|---|
| タイトル | Z-DNA HEXAMER WITH 5' OVERHANGS THAT FORM A REVERSE WATSON-CRICK BASE PAIR |
|---|
 要素 要素 | - DNA (5'-D(*CP*CP*GP*CP*GP*CP*G)-3')
- DNA (5'-D(*GP*CP*GP*CP*GP*CP*G)-3')
|
|---|
 キーワード キーワード | DNA / Z-DNA / DOUBLE HELIX / OVERHANGING BASE / FLIPPED-OUT BASE |
|---|
| 機能・相同性 | COBALT HEXAMMINE(III) / DNA 機能・相同性情報 機能・相同性情報 |
|---|
| 手法 |  X線回折 / 分子置換 / 解像度: 1.8 Å X線回折 / 分子置換 / 解像度: 1.8 Å |
|---|
 データ登録者 データ登録者 | Mooers, B.H.M. / Eichman, B.F. / Ho, P.S. |
|---|
 引用 引用 | ジャーナル: J.Mol.Biol. / 年: 1997
タイトル: The structures and relative stabilities of d(G x G) reverse Hoogsteen, d(G x T) reverse wobble, and d(G x C) reverse Watson-Crick base-pairs in DNA crystals.
著者: Mooers, B.H. / Eichman, B.F. / Ho, P.S. |
|---|
| 履歴 | | 登録 | 1997年2月4日 | 登録サイト: NDB / 処理サイト: NDB |
|---|
| 改定 1.0 | 1997年8月28日 | Provider: repository / タイプ: Initial release |
|---|
| 改定 1.1 | 2008年5月22日 | Group: Version format compliance |
|---|
| 改定 1.2 | 2011年7月13日 | Group: Version format compliance |
|---|
| 改定 1.3 | 2024年2月21日 | Group: Data collection / Database references / Derived calculations
カテゴリ: chem_comp_atom / chem_comp_bond ...chem_comp_atom / chem_comp_bond / database_2 / pdbx_struct_conn_angle / struct_conn / struct_site
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession ..._database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr2_auth_seq_id / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id |
|---|
| 改定 1.4 | 2024年4月3日 | Group: Refinement description / カテゴリ: pdbx_initial_refinement_model |
|---|
|
|---|
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード







 PDBj
PDBj


 集合体
集合体

 分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg
分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg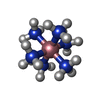
 分子量: 161.116 Da / 分子数: 1 / 由来タイプ: 合成 / 式: CoH18N6
分子量: 161.116 Da / 分子数: 1 / 由来タイプ: 合成 / 式: CoH18N6 分子量: 18.015 Da / 分子数: 69 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 69 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析