THE POLYPEPTIDE CHAIN CONTAINS THREE (3) COPIES OF THE TRAP PROTEIN LINKED IN TANDEM, WHICH ARRANGE THEMSELVES TO MAKE A 12-MER RING IN SOLUTION. EACH CHAIN IN THIS MODEL REPRESENTS ONE COPY OF TRAP, NOT A SEPARATE POLYPEPTIDE. THE LINKER PEPTIDES ARE MAINLY NOT VISIBLE IN THE ELECTRON DENSITY. THE 12MER RINGS ARE ALIGNED WITH THE CRYSTALLOGRAPHIC FOUR-FOLD AXIS. THERE ARE SIX COPIES OF TRAP PRESENT IN THE ASYMMETRIC UNIT. FOR THIS PROTEIN, CALLED T3A7, THE LINKER PEPTIDES CONSIST OF SEVEN (7) ALANINE RESIDUES.
-
要素
#1: タンパク質
TranscriptionattenuationproteinmtrB / Tryptophan RNA-binding attenuator protein / Trp RNA-binding attenuation protein / TRAP
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 Geobacillus stearothermophilus (バクテリア)
Geobacillus stearothermophilus (バクテリア) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード













 PDBj
PDBj
 集合体
集合体


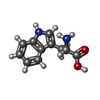
 タイプ: L-peptide linking / 分子量: 204.225 Da / 分子数: 6 / 由来タイプ: 合成 / 式: C11H12N2O2
タイプ: L-peptide linking / 分子量: 204.225 Da / 分子数: 6 / 由来タイプ: 合成 / 式: C11H12N2O2 分子量: 18.015 Da / 分子数: 201 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 201 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: BL-17A / 波長: 1 Å
/ ビームライン: BL-17A / 波長: 1 Å 解析
解析