 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-2zd0: Crystal structures and thermostability of mutant TRAP3 A5 (ENGINE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2zd0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structures and thermostability of mutant TRAP3 A5 (ENGINEERED TRAP) | ||||||
 Components Components | Transcription attenuation protein mtrB | ||||||
 Keywords Keywords | RNA BINDING PROTEIN / LINKER / ARTIFICIAL / ENGINEERED / RING PROTEIN / 12-mer | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA-templated transcription termination / regulation of DNA-templated transcription / RNA binding Similarity search - Function | ||||||
| Biological species |   Geobacillus stearothermophilus (bacteria) Geobacillus stearothermophilus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Watanabe, M. / Mishima, Y. / Yamashita, I. / Park, S.Y. / Tame, J.R.H. / Heddle, J.G. | ||||||
 Citation Citation | Journal: Protein Sci. / Year: 2008 Title: Intersubunit linker length as a modifier of protein stability: crystal structures and thermostability of mutant TRAP. Authors: Watanabe, M. / Mishima, Y. / Yamashita, I. / Park, S.Y. / Tame, J.R. / Heddle, J.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2zd0.cif.gz | 51.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2zd0.ent.gz | 37.3 KB | Display | PDB format |
| PDBx/mmJSON format | 2zd0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zd/2zd0ftp://data.pdbj.org/pub/pdb/validation_reports/zd/2zd0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2zczC  2exsS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | THE POLYPEPTIDE CHAIN CONTAINS THREE (3) COPIES OF THE TRAP PROTEIN LINKED IN TANDEM, WHICH ARRANGE THEMSELVES TO MAKE A 12-MER RING IN SOLUTION. EACH CHAIN IN THIS MODEL REPRESENTS ONE COPY OF TRAP, NOT A SEPARATE POLYPEPTIDE. THE LINKER PEPTIDES ARE NOT VISIBLE IN THE ELECTRON DENSITY. THE 12MER RINGS ARE ALIGNED WITH THE CRYSTALLOGRAPHIC FOUR-FOLD AXIS. THERE ARE THREE COPIES OF TRAP PRESENT IN THE ASYMMETRIC UNIT. FOR THIS PROTEIN, CALLED T3A5, THE LINKER PEPTIDES CONSIST OF FIVE (5) ALANINE RESIDUES. |
-Components
| #1: Protein | Mass: 8612.765 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Geobacillus stearothermophilus (bacteria)Strain: NCA 26, ATCC 12980 / Gene: mtrB / Plasmid: pET21b / Species (production host): Escherichia coli / Production host: #2: Chemical | 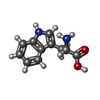  Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C11H12N2O2 Type: L-peptide linking / Mass: 204.225 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C11H12N2O2#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 39 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 39 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.14 Å3/Da / Density % sol: 42.49 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 10.5 Details: 0.09M CAPS pH 10.5, 30%(w/v) PEG300, 0.15M Ammonium sulfate, 10mM L-tryptophan, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Photon Factory  / Beamline: AR-NW12A / Wavelength: 1 Å / Beamline: AR-NW12A / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Jun 7, 2006 / Details: mirrors |
| Radiation | Monochromator: Si / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.5→50 Å / Num. obs: 7898 / % possible obs: 95.7 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 5 % / Biso Wilson estimate: 27.4 Å2 / Rmerge(I) obs: 0.061 / Rsym value: 0.048 / Net I/σ(I): 12.3 |
| Reflection shell | Resolution: 2.5→2.59 Å / Redundancy: 4.1 % / Rmerge(I) obs: 0.221 / Mean I/σ(I) obs: 11.7 / Num. unique all: 744 / Rsym value: 0.246 / % possible all: 93.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2EXS Resolution: 2.5→20 Å / Cor.coef. Fo:Fc: 0.934 / Cor.coef. Fo:Fc free: 0.899 / SU B: 7.967 / SU ML: 0.185 / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.609 / ESU R Free: 0.301 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 27.491 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.5→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.497→2.561 Å / Rfactor Rfree error: 0.08 / Total num. of bins used: 20
|
