 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2yet | ||||||
|---|---|---|---|---|---|---|---|
| Title | Thermoascus GH61 isozyme A | ||||||
 Components Components | GH61 ISOZYME A | ||||||
 Keywords Keywords | HYDROLASE / DEGRADATION OF BIOMASS | ||||||
| Function / homology |  Function and homology information Function and homology informationlytic cellulose monooxygenase (C1-hydroxylating) / lytic cellulose monooxygenase (C4-dehydrogenating) / cellulose catabolic process / monooxygenase activity / extracellular region / metal ion binding Similarity search - Function | ||||||
| Biological species |  THERMOASCUS AURANTIACUS (fungus) THERMOASCUS AURANTIACUS (fungus) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.502 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.502 Å | ||||||
 Authors Authors | Otten, H. / Quinlan, R.J. / Sweeney, M.D. / Poulsen, J.-C.N. / Johansen, K.S. / Krogh, K.B.R.M. / Joergensen, C.I. / Tovborg, M. / Anthonsen, A. / Tryfona, T. ...Otten, H. / Quinlan, R.J. / Sweeney, M.D. / Poulsen, J.-C.N. / Johansen, K.S. / Krogh, K.B.R.M. / Joergensen, C.I. / Tovborg, M. / Anthonsen, A. / Tryfona, T. / Walter, C.P. / Dupree, P. / Xu, F. / Davies, G.J. / Walton, P.H. / Lo Leggio, L. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2011 Title: Insights Into the Oxidative Degradation of Cellulose by a Copper Metalloenzyme that Exploits Biomass Components. Authors: Quinlan, R.J. / Sweeney, M.D. / Lo Leggio, L. / Otten, H. / Poulsen, J.-C.N. / Johansen, K.S. / Krogh, K.B.R.M. / Jorgensen, C.I. / Tovborg, M. / Anthonsen, A. / Tryfona, T. / Walter, C.P. / ...Authors: Quinlan, R.J. / Sweeney, M.D. / Lo Leggio, L. / Otten, H. / Poulsen, J.-C.N. / Johansen, K.S. / Krogh, K.B.R.M. / Jorgensen, C.I. / Tovborg, M. / Anthonsen, A. / Tryfona, T. / Walter, C.P. / Dupree, P. / Xu, F. / Davies, G.J. / Walton, P.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2yet.cif.gz | 202.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2yet.ent.gz | 161.4 KB | Display | PDB format |
| PDBx/mmJSON format | 2yet.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ye/2yetftp://data.pdbj.org/pub/pdb/validation_reports/ye/2yet | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 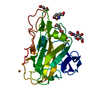 3zudC  2vtcS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||
| 2 | 
| ||||||||||||||||||
| Unit cell |
| ||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments:
NCS oper: (Code: given Matrix: (1, 0.004824, -0.00431), Vector: |
-Components
-Protein / Sugars , 2 types, 3 molecules AB
| #1: Protein | Mass: 24418.043 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Details: GH61 FRAGMENT. METHYLATION AT 4-N-H1, AND TWO CYSTINES AT C56-178 AND C97-C101 IN BOTH PROTEIN CHAINS. N-ACETYLGLUCOSAMIN-GLYCOSYLATION AT N138 IN THE B CHAIN AND NOT CLEARLY VISIBLE IN CHAIN A. Source: (gene. exp.) THERMOASCUS AURANTIACUS (fungus) / Production host: #4: Sugar | ChemComp-NAG / |  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C8H15NO6 |
|---|
-Non-polymers , 4 types, 632 molecules 



| #2: Chemical |  Mass: 63.546 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cu Mass: 63.546 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cu#3: Chemical | ChemComp-ACE /  Mass: 44.053 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H4O Mass: 44.053 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C2H4O#5: Chemical | ChemComp-GOL / |  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 625 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Sequence details | LACKING THE 21 AMINO ACID SIGNAL PEPTIDE GENBANK: ACS05720.1 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.92 Å3/Da / Density % sol: 35.44 % / Description: NONE |
|---|---|
| Crystal grow | pH: 8 Details: 0.2 M NACL, 0.1 M HEPES, PH 8.0 AND 25%(W/V) PEG 3350. CRYO PROTECTED WITH 8%(W/V) GLYCEROL, 8%(W/V) ETHYLENE GLYCOL, 9%(W/V)D-SUCROSE AND 2%(W/V)D-GLUCOSE |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: MAX II  / Beamline: I911-2 / Wavelength: 1.0379 / Beamline: I911-2 / Wavelength: 1.0379 |
| Detector | Type: MARRESEARCH SX-165 / Detector: CCD / Date: Jun 28, 2008 Details: MULTILAYER MIRROR, CURVED TO FOCUS IN THE VERTICAL (R IS 400 M) |
| Radiation | Monochromator: BENT SI (111) CRYSTAL, HORIZONTALLY FOCUSING / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0379 Å / Relative weight: 1 |
| Reflection | Resolution: 1.49→12.33 Å / Num. obs: 67162 / % possible obs: 93.2 % / Observed criterion σ(I): 0 / Redundancy: 6.1 % / Biso Wilson estimate: 12.08 Å2 / Rmerge(I) obs: 0.07 / Net I/σ(I): 16.3 |
| Reflection shell | Resolution: 1.49→1.57 Å / Redundancy: 5.2 % / Rmerge(I) obs: 0.45 / Mean I/σ(I) obs: 3.3 / % possible all: 74.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 2VTC Resolution: 1.502→12.333 Å / SU ML: 0.18 / σ(F): 0 / Phase error: 18.72 / Stereochemistry target values: ML Details: RESIDUES 25-27 ARE DISORDERED IN BOTH CHAINS. THERE IS WELL DEFINED ELECTRON DENSITY FOR THE RAMACHANDRAN OUTLIERS H57 IN BOTH CHAINS. DISORDERED LOOP 25-27, MODELED IN TWO CONFORMATIONS FOR ...Details: RESIDUES 25-27 ARE DISORDERED IN BOTH CHAINS. THERE IS WELL DEFINED ELECTRON DENSITY FOR THE RAMACHANDRAN OUTLIERS H57 IN BOTH CHAINS. DISORDERED LOOP 25-27, MODELED IN TWO CONFORMATIONS FOR THE B CHAIN, GLYCOSYLATION JUST IN B CHAIN
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0 Å / VDW probe radii: 0.3 Å / Solvent model: FLAT BULK SOLVENT MODEL / Bsol: 87.95 Å2 / ksol: 0.556 e/Å3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.3 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.502→12.333 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
