Mass: 18.015 Da / Num. of mol.: 274 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.04 Å3/Da / Density % sol: 39.83 % / Description: NONE
Crystal grow
Temperature: 293 K / Method: vapor diffusion, sitting drop Details: SITTING-DROP VAPOR-DIFFUSION METHOD AT 293 K. DROPS WITH 300 NANOLITERS PROTEIN SOLUTION (PROTEIN AT 40 MG/ML IN 20 MM BIS-TRIS-PROPANE, PH 6.5, 0.1 M NACL WITH 10 MM CMP, 10 MM MGCL2 AND 10 ...Details: SITTING-DROP VAPOR-DIFFUSION METHOD AT 293 K. DROPS WITH 300 NANOLITERS PROTEIN SOLUTION (PROTEIN AT 40 MG/ML IN 20 MM BIS-TRIS-PROPANE, PH 6.5, 0.1 M NACL WITH 10 MM CMP, 10 MM MGCL2 AND 10 MM ERYTHRITOL) AND 300 NANOLITERS SCREENING/RESERVOIR SOLUTION (10% (W/V) PEG 20,000, 20% (V/V) MONOMETHYL ETHER PEG 550, 0.03M NAF, 0.03M NAI, 0.03M NABR, AND 0.1M MOPS/NA-HEPES, PH 7.5)
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information MYCOBACTERIUM SMEGMATIS (bacteria)
MYCOBACTERIUM SMEGMATIS (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj

 Assembly
Assembly
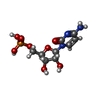
 Mass: 323.197 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H14N3O8P
Mass: 323.197 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H14N3O8P Mass: 18.015 Da / Num. of mol.: 274 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 274 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID14-2 / Wavelength: 0.933
/ Beamline: ID14-2 / Wavelength: 0.933  Processing
Processing